A Closer Look into the Role of Protein Tau in the Identification of Promising Therapeutic Targets for Alzheimer’s Disease


Abstract
1. Introduction
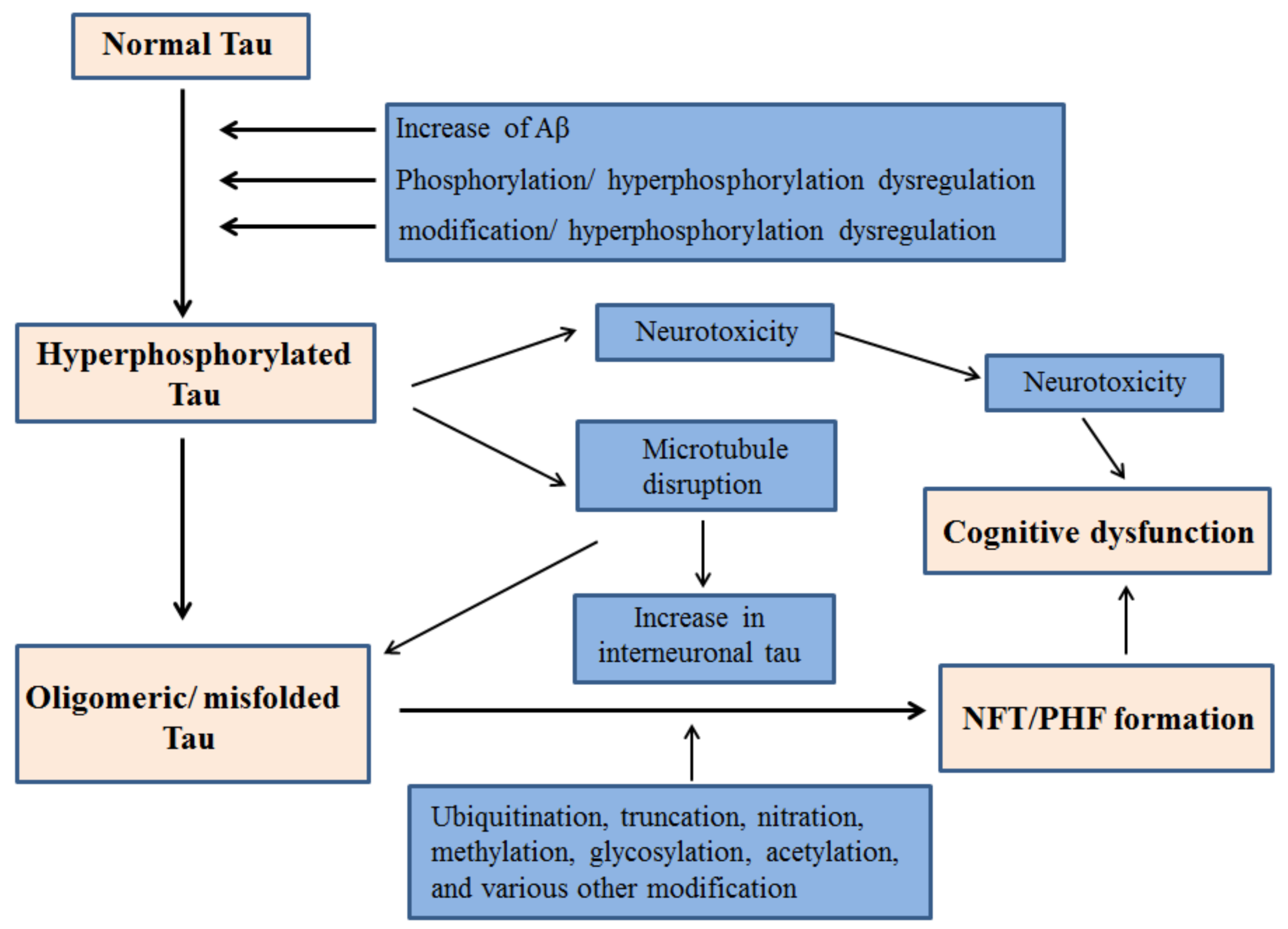
2. Tau Hyperphosphorylation in AD
2.1. GSK-3β Inhibition in Tau
2.2. Connection between Tau Hyperphosphorylation and Aβ
2.3. Aβ-Facilitated Increase in Tau phosphorylation in Animal Models
3. Tau Mediated Neurotoxicity, Secretion and Inter-Cellular Transfer
3.1. Neurotoxicity from Tau
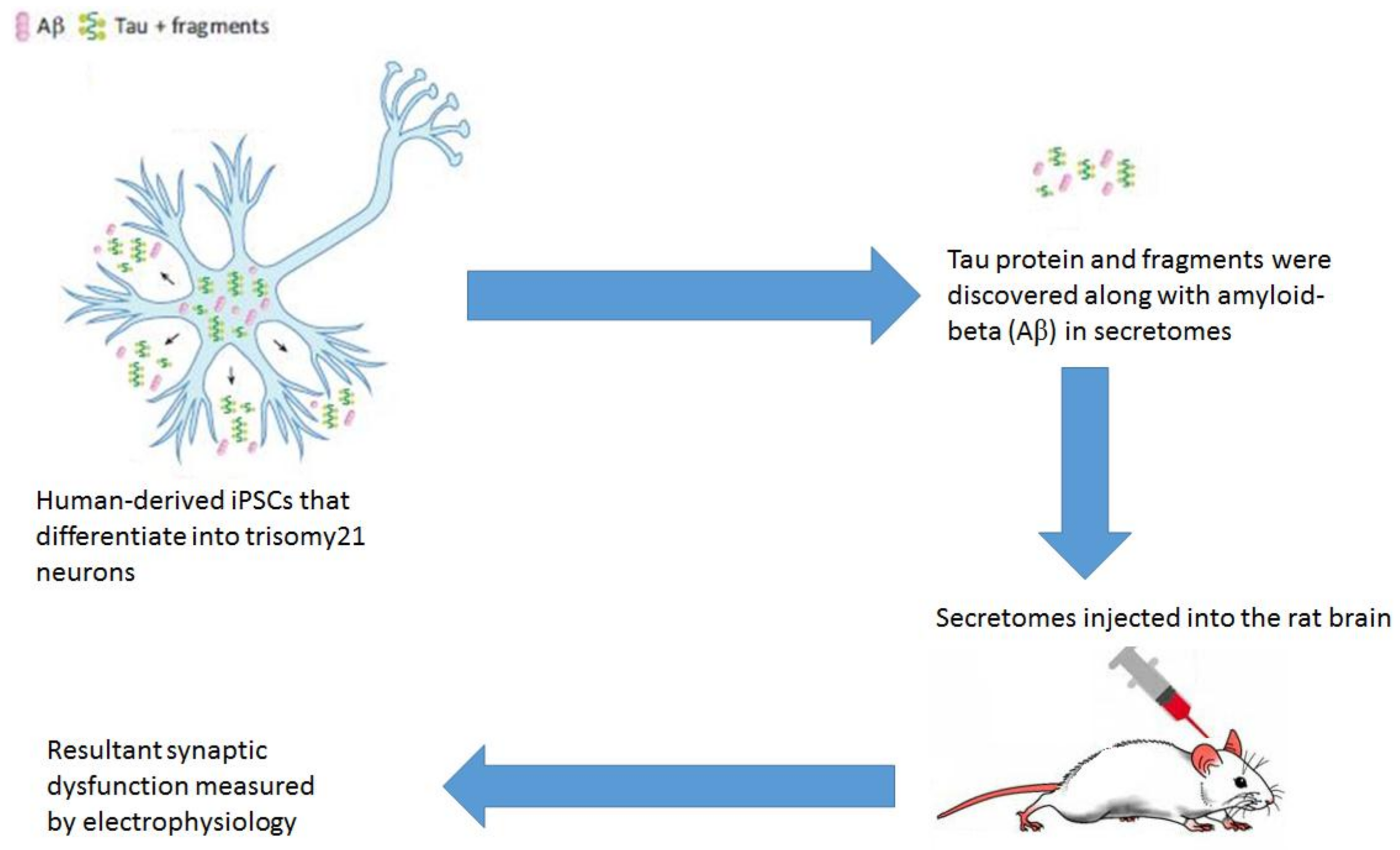
3.2. Tau Secretion
3.3. Tau Inter-Cellular Transfer
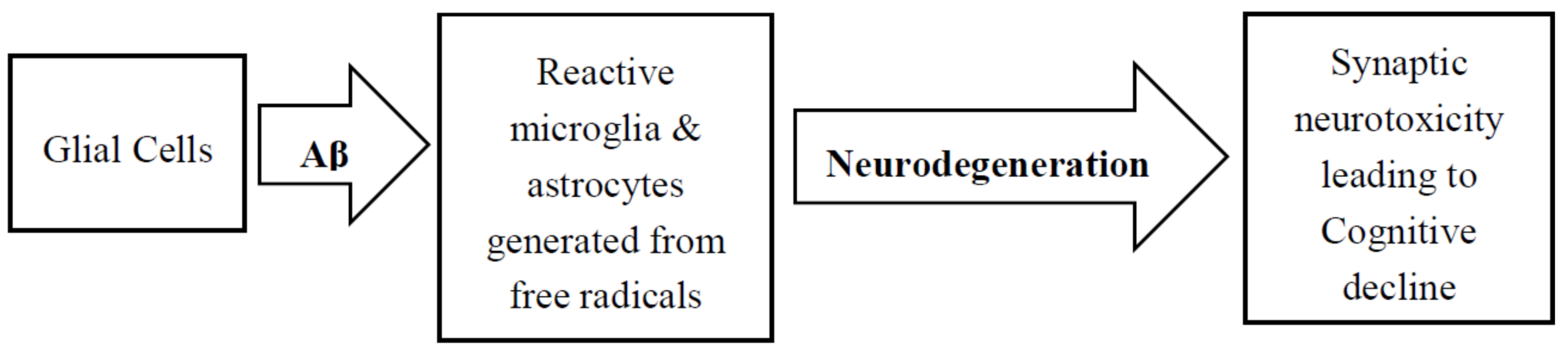
4. Role of Glial Cells in AD Pathology
5. Diagnostic Approaches for AD Using Tau-Imaging
6. Immune Responses and Neuroprotection in Tau Pathology: Therapeutic Opportunities
6.1. Immune Responses and Neuroinflammation
6.2. Neuroprotection against AD-Tau
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Federico, C.; Gil, L.; Bruno, F.; D’Amico, A.G.; D’Agata, V.; Saccone, S. Phosphorylated nucleolar Tau protein is related to the neuronal in vitro differentiation. Gene 2018, 664, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; Wischik, C.; Hof, P. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Raviv, U.; Miller, H.P.; Gaylord, M.R.; Kiris, E.; Ventimiglia, D.; Needleman, D.J.; Kim, M.W.; Wilson, L.; Feinstein, S.C.; et al. Human Microtubule-Associated-Protein Tau Regulates the Number of Protofilaments in Microtubules: A Synchrotron X-Ray Scattering Study. Biophys. J. 2009, 97, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Kaluski, S.; Portillo, M.; Besnard, A.; Mostoslavsky, R.; Sahay, A.; Correspondence, D.T.; Stein, D.; Einav, M.; Zhong, L.; Ueberham, U.; et al. Neuroprotective Functions for the Histone Deacetylase SIRT6 Cell Reports Report Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 2017, 18, 3052–3062. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Ferrer, I.; Grinberg, L.T.; Alafuzoff, I.; Attems, J.; Budka, H.; Cairns, N.J.; Crary, J.F.; Duyckaerts, C.; Ghetti, B.; et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016, 131, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lante, F.; Buisson, A. Activity-Dependent Tau Protein Translocation to Excitatory Synapse Is Disrupted by Exposure to Amyloid-Beta Oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. B Biol. Sci. 2013, 369, 20130144. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [PubMed]
- Tai, H.-C.; Wang, B.Y.; Serrano-Pozo, A.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 146. [Google Scholar] [CrossRef] [PubMed]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Chouala, M.S.; Sultan, A.; Marciniak, E.; Humez, S.; Binder, L.; Kayed, R.; et al. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis. 2015, 82, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Seereeram, A.; Noble, W. Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.-L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev. Proteomics 2008, 5, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Papon, M.-A.; El Khoury, N.B.; Marcouiller, F.; Julien, C.; Morin, F.; Bretteville, A.; Petry, F.R.; Gaudreau, S.; Amrani, A.; Mathews, P.M.; et al. Deregulation of protein phosphatase 2A and hyperphosphorylation of τ protein following onset of diabetes in NOD mice. Diabetes 2013, 62, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Zheng-Fischhöfer, Q.; Biernat, J.; Mandelkow, E.M.; Illenberger, S.; Godemann, R.; Mandelkow, E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998, 252, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Malia, T.J.; Teplyakov, A.; Ernst, R.; Wu, S.-J.; Lacy, E.R.; Liu, X.; Vandermeeren, M.; Mercken, M.; Luo, J.; Sweet, R.W.; Gilliland, G.L. Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins Struct. Funct. Bioinforma. 2016, 84, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. J. Alzheimer’s Dis. 2012, 33, S123–S139. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.d.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Stanic, G.; Mladinov, M.; Jovanov-Milosevic, N.; Kostovic, I.; Hof, P.R. Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 2009, 35, 532–554. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta - Mol. Basis Dis. 2016, 1862, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in Neurodegenerative Diseases. Int. J. Alzheimers. Dis. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jembrek, M.; Babić, M.; Pivac, N.; Hof, P.; Šimić, G. Hyperphosphorylation of tau by GSK-3β in Alzheimer’s disease: The interaction of Aβ and sphingolipid mediators as a therapeutic target. Transl. Neurosci. 2013, 4, 466–476. [Google Scholar] [CrossRef]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K.; Imahori, K. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef]
- Bijur, G.N.; Jope, R.S. Proapoptotic Stimuli Induce Nuclear Accumulation of Glycogen Synthase Kinase-3β. J. Biol. Chem. 2001, 276, 37436–37442. [Google Scholar] [CrossRef] [PubMed]
- Lace, G.; Savva, G.M.; Forster, G.; de Silva, R.; Brayne, C.; Matthews, F.E.; Barclay, J.J.; Dakin, L.; Ince, P.G.; Wharton, S.B. MRC-CFAS Hippocampal tau pathology is related to neuroanatomical connections: An ageing population-based study. Brain 2009, 132, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Stratmann, K.; Heinsen, H.; Del Turco, D.; Ghebremedhin, E.; Seidel, K.; den Dunnen, W.; Korf, H.-W. Hierarchical Distribution of the Tau Cytoskeletal Pathology in the Thalamus of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2015, 49, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007, 25, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.W.; Spreen, R.C.; Herman, J.L.; Chow, F.P.; Davison, M.D.; Young, J.; Caputo, C.B. Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J. Biol. Chem. 1993, 268, 1166–1173. [Google Scholar] [PubMed]
- Litersky, J.M.; Johnson, G.V.; Jakes, R.; Goedert, M.; Lee, M.; Seubert, P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem. J. 1996, 316, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Sironi, J.J.; Yen, S.H.; Gondal, J.A.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 1998, 436, 471–475. [Google Scholar] [CrossRef]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation that Detaches Tau Protein from Microtubules (Ser262, Ser214) Also Protects It against Aggregation into Alzheimer Paired Helical Filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; Liu, R.; Wang, Q.; Grundke-Iqbal, I.; Iqbal, K. In vitro analysis of tau phosphorylation sites and its biological activity. Chinese Med. Sci. J. Chung-kuo i hsueh k’o hsueh tsa chih 2002, 17, 13–16. [Google Scholar]
- Ksiezak-Reding, H.; Pyo, H.K.; Feinstein, B.; Pasinetti, G.M. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro. Biochim. Biophys. Acta 2003, 1639, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Kawamata, T.; Mukai, H.; Hasegawa, H.; Isagawa, T.; Yasuda, M.; Hashimoto, T.; Terashima, A.; Nakai, M.; Ono, Y.; Tanaka, C.; Tanaka, C. Phosphorylation of Tau Is Regulated by PKN. J. Biol. Chem. 2001, 276, 10025–10031. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Grundke-Iqbal, I.; Iqbal, K.; Bogdanovic, N.; Winblad, B.; Cowburn, R.F. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 1998, 797, 267–277. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Hartigan, J.A. Tau protein in normal and Alzheimer’s disease brain: an update. J. Alzheimers. Dis. 1999, 1, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Godemann, R.; Biernat, J.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation of tau protein by recombinant GSK-3β: Pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999, 454, 157–164. [Google Scholar] [CrossRef]
- Wang, J.Z.; Wu, Q.; Smith, A.; Grundke-Iqbal, I.; Iqbal, K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998, 436, 28–34. [Google Scholar] [CrossRef]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of Pathological Tau Species and Memory Loss in a Conditional Model of Tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.-M.; Müller, D.J.; Mandelkow, E. Oligomer Formation of Tau Protein Hyperphosphorylated in Cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.d.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.-S.; Freeman, J.; Rebeck, G.W. Apolipoprotein E decreases tau kinases and phospho-tau levels in primary neurons. Mol. Neurodegener. 2006, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Duff, K. Linking Abeta and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, X.Q.; Wyss-Coray, T.; Brecht, W.J.; Sanan, D.A.; Mahley, R.W. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 8838–8843. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, M.; Braithwaite, S.P.; Stock, J.B. Phosphoprotein phosphatase 2A: A novel druggable target for Alzheimer’s disease. Future Med. Chem. 2011, 3, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-W.; Gustafsson, J.-Å.; Tanila, H.; Bjorkdahl, C.; Liu, R.; Winblad, B.; Pei, J.-J. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol. Dis. 2008, 31, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Xing, Y.; Gong, L.; Li, H.; Wu, Z.; Li, Y.; Wang, J.; Wang, Y.; Dong, L.; Li, S. Effects of ginsenoside Rg1 or 17β-estradiol on a cognitively impaired, ovariectomized rat model of Alzheimer’s disease. Neuroscience 2012, 220, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-P.; Zhang, Y.; Yao, X.-Q.; Zhang, C.-E.; Fang, J.; Wang, Q.; Wang, J.-Z. Activation of glycogen synthase kinase-3 inhibits protein phosphatase-2A and the underlying mechanisms. Neurobiol. Aging 2008, 29, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.A.; Ziburkus, J.; Jankowsky, J.; Rasband, M.N. Amyloid-β plaques disrupt axon initial segments. Exp. Neurol. 2016, 281, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell. Longev. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau. JAMA Neurol. 2014, 71, 505. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.-C.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol. Neurodegener. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Nery, L.R.; Eltz, N.S.; Hackman, C.; Fonseca, R.; Altenhofen, S.; Guerra, H.N.; Freitas, V.M.; Bonan, C.D.; Vianna, M.R.M.R. Brain Intraventricular Injection of Amyloid-β in Zebrafish Embryo Impairs Cognition and Increases Tau Phosphorylation, Effects Reversed by Lithium. PLoS ONE 2014, 9, e105862. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Jiang, Z.-F. Accumulated Amyloid-β Peptide and Hyperphosphorylated Tau Protein: Relationship and Links in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Rosen, K.M.; Smith, R.C.; Walsh, K.; Gouras, G.K.; Querfurth, H.W. Intraneuronal -Amyloid Expression Downregulates the Akt Survival Pathway and Blunts the Stress Response. J. Neurosci. 2005, 25, 10960–10969. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.-M. A Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.D.; Ermak, G.; Davies, K.J.A. RCAN1-1L is overexpressed in neurons of Alzheimer’s disease patients. FEBS J. 2007. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.; Levenga, J.; Cain, P.; Rothermel, B.; Klann, E.; Hoeffer, C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2015, 130, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.-C.; Giraldo, E.; Ermak, G.; Alonso, M.-D.; Pallardó, F.V.; Davies, K.J.A.; Viña, J. Amyloid-β Toxicity and Tau Hyperphosphorylation are Linked Via RCAN1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Porta, S.; Serra, S.A.; Huch, M.; Valverde, M.A.; Llorens, F.; Estivill, X.; Arbonés, M.L.; Martí, E. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: A potential pathogenic process in neurodegeneration. Hum. Mol. Genet. 2007, 16, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Ferreiro, E.; Pereira, C.; Oliveira, C.R. ER stress is involved in Aβ-induced GSK-3β activation and tau phosphorylation. J. Neurosci. Res. 2008, 86, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, M.H.; Carvalho, M.M.V.F.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β Binds to the Extracellular Cysteine-rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Gladbach, A.; Pennanen, L.; van Eersel, J.; Schild, A.; David, D.; Ittner, L.M. Animal models reveal role for tau phosphorylation in human disease. Biochim. Biophys. Acta - Mol. Basis Dis. 2010, 1802, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sintes, R.; Hernández, F.; Lucas, J.J.; Avila, J. GSK-3 Mouse Models to Study Neuronal Apoptosis and Neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, M.A.; Blurton-Jones, M.; Agazaryan, A.A.; Nerhus, J.L.; Martinez-Coria, H.; LaFerla, F.M. Soluble A Promotes Wild-Type Tau Pathology In vivo. J. Neurosci. 2012, 32, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Carreras, I.; Hossain, L.; Ryu, H.; Klein, W.L.; Oddo, S.; LaFerla, F.M.; Jenkins, B.G.; Kowall, N.W.; Dedeoglu, A. Ibuprofen reduces Aβ, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008, 1207, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Sarnico, I.; Benarese, M.; Branca, C.; Baiguera, C.; Hutter-Paier, B.; Windisch, M.; Spano, P.; Imbimbo, B.P.; Pizzi, M. The γ-Secretase Modulator CHF5074 Reduces the Accumulation of Native Hyperphosphorylated Tau in a Transgenic Mouse Model of Alzheimer’s Disease. J. Mol. Neurosci. 2011, 45, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.-C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; Cho, K. Aβ1–42 inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Bennecib, M.; Grignon, Y.; Uchihara, T.; He, Y.; Piette, F.; Hauw, J.J. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol. Aging 1997, 18, 267–273. [Google Scholar] [CrossRef]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, M.J.; Devous, M.D.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F.W.; Jennings, D.; Arora, A.K.; McGeehan, A.; Lim, N.C.; et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 2017, 140, 334. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [PubMed]
- Venero, J.L.; Burguillos, M.A.; Joseph, B. Caspases Playing in the Field of Neuroinflammation: Old and New Players. Dev. Neurosci. 2013, 35, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Brück, W.; Jellinger, K.; Lassmann, H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef]
- Rohn, T.T.; Head, E.; Nesse, W.H.; Cotman, C.W.; Cribbs, D.H. Activation of Caspase-8 in the Alzheimer’s Disease Brain. Neurobiol. Dis. 2001, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Head, E.; Su, J.H.; Anderson, A.J.; Bahr, B.A.; Cotman, C.W.; Cribbs, D.H. Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer’s disease. Am. J. Pathol. 2001, 158, 189–198. [Google Scholar] [CrossRef]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001, 898, 350–357. [Google Scholar] [CrossRef]
- Rohn, T.T.; Rissman, R.A.; Davis, M.C.; Kim, Y.E.; Cotman, C.W.; Head, E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Gastard, M.C.; Troncoso, J.C.; Koliatsos, V.E. Caspase activation in the limbic cortex of subjects with early Alzheimer’s disease. Ann. Neurol. 2003, 54, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.-G.; Liao, X.-M.; Chen, X.-Q.; Liu, G.-P.; Wang, J.-Z. Effects of tau phosphorylation on proteasome activity. FEBS Lett. 2007, 581, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Wang, H.; Wang, G. The ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2013, 19, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Hall, G.F.; Saman, S. Death or secretion? The demise of a plausible assumption about CSF-tau in Alzheimer Disease? Commun. Integr. Biol. 2012, 5, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Avila, J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 2014, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, T.T.; Koivisto, A.M.; Hartikainen, P.; Helisalmi, S.; Soininen, H.; Herukka, S.-K. Longitudinal Changes of CSF Biomarkers in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 25, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.-M.; Diamond, M.I.; Lee, V.M.-Y.; Holtzman, D.M. In vivo Microdialysis Reveals Age-Dependent Decrease of Brain Interstitial Fluid Tau Levels in P301S Human Tau Transgenic Mice. J. Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, V.; Mohamed, N.-V.; Rivest-McGraw, J.; Bertrand, J.; Lauzon, M.; Leclerc, N. Hyperphosphorylation and Cleavage at D421 Enhance Tau Secretion. PLoS ONE 2012, 7, e36873. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; Hall, G.F. Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; Mandelkow, E.-M. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.-L.B.; Davtyan, H.; Housley, S.B.; Blair, L.J.; Gillies, A.; Nordhues, B.A.; Zhang, B.; Liu, J.; Gestwicki, J.E.; Lee, D.C.; Gordon, M.N.; Morgan, D.; Dickey, C.A. Epitope analysis following active immunization with tau proteins reveals immunogens implicated in tau pathogenesis. J. Neuroinflammation 2014, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Doran, E.; Keator, D.; Head, E.; Phelan, M.J.; Kim, R.; Totoiu, M.; Barrio, J.R.; Small, G.W.; Potkin, S.G.; Lott, I.T. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J. Alzheimer’s Dis. 2017, 56, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.-W.; Corbett, G.T.; Moore, S.; Klyubin, I.; O’Malley, T.T.; Walsh, D.M.; Livesey, F.J.; Rowan, M.J. Extracellular Forms of Aβ and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep. 2018, 23, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B. The Way of Tau: Secretion and Synaptic Dysfunction. Trends Mol. Med. 2018, 24, 595–597. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; Falcon, B.; Vaysburd, M.; Clift, D.; Oblak, A.L.; Ghetti, B.; Goedert, M.; James, L.C. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Wadia, J.S.; Zhu, X.; Keogh, E.; Kükrer, B.; van Ameijde, J.; Inganäs, H.; Siregar, B.; Perdok, G.; Diefenbach, O.; et al. Immunological memory to hyperphosphorylated tau in asymptomatic individuals. Acta Neuropathol. 2017, 133, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Agadjanyan, M.G.; Petrovsky, N.; Ghochikyan, A. A fresh perspective from immunologists and vaccine researchers: Active vaccination strategies to prevent and reverse Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; David, J.P.; Sergeant, N.; Buée, L.; Wattez, A.; Vermersch, P.; Ghozali, F.; Fallet-Bianco, C.; Pasquier, F.; Lebert, F.; et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999, 52, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Grober, E.; Dickson, D.; Sliwinski, M.J.; Buschke, H.; Katz, M.; Crystal, H.; Lipton, R.B. Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 1999, 20, 573–579. [Google Scholar] [CrossRef]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.-C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef] [PubMed]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L.; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Dorothée, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Légeron, F.-P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.S.; Lal, D.; Zhang, B.; Dabir, D.V.; Swanson, E.; Lee, V.M.-Y.; Trojanowski, J.Q. Transgenic Mouse Model of Tau Pathology in Astrocytes Leading to Nervous System Degeneration. J. Neurosci. 2005, 25, 3539–3550. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in vivo. Science (80-. ). 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.-È. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.-F. Understanding the neurobiology of CD200 and the CD200 receptor: A therapeutic target for controlling inflammation in human brains? Future Neurol. 2013, 8, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S. Neuron-Microglia Interactions in Mental Health Disorders: “For Better, and For Worse”. Front. Immunol. 2016, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.-V.; Doty, K.R.; Town, T. Innate Immunity Fights Alzheimer’s Disease. Trends Neurosci. 2015, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 349. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Nadrigny, F.; Regen, T.; Martinez-Hernandez, A.; Dumitrescu-Ozimek, L.; Terwel, D.; Jardanhazi-Kurutz, D.; Walter, J.; Kirchhoff, F.; Hanisch, U.-K.; et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA 2010, 107, 6058–6063. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Johnson, M.F.; Kobritz, N.; Eslami, P.; Poteshkina, A.; Varadarajan, S.; Turman, J.; Zemlan, F.; Harris-White, M.E. Oral TNFα Modulation Alters Neutrophil Infiltration, Improves Cognition and Diminishes Tau and Amyloid Pathology in the 3xTgAD Mouse Model. PLoS ONE 2015, 10, e0137305. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-ike pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Naert, G.; Rivest, S. CC Chemokine Receptor 2 Deficiency Aggravates Cognitive Impairments and Amyloid Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 6208–6220. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Kim, D.; Katsumoto, A.; Kokiko-Cochran, O.N.; Lamb, B.T.; Ransohoff, R.M. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J. Neuroinflammation 2015, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Schraen-Maschke, S.; Sergeant, N.; Dhaenens, C.-M.; Bombois, S.; Deramecourt, V.; Caillet-Boudin, M.-L.; Pasquier, F.; Maurage, C.-A.; Sablonnière, B.; Vanmechelen, E.; et al. Tau as a biomarker of neurodegenerative diseases. Biomark. Med. 2008, 2, 363–384. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.H.; Herukka, S.-K.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S.; et al. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimer’s Dement. 2017, 13, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.-Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica (Cairo). 2012, 2012, 796024. [Google Scholar] [CrossRef] [PubMed]
- Beckett, L.A.; Harvey, D.J.; Gamst, A.; Donohue, M.; Kornak, J.; Zhang, H.; Kuo, J.H.; Alzheimer’s Disease Neuroimaging Initiative. The Alzheimer’s Disease Neuroimaging Initiative: Annual change in biomarkers and clinical outcomes. Alzheimers. Dement. 2010, 6, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Ukoumunne, O.; Ladds, E.C.; Martin, S. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2017, 3, CD010803. [Google Scholar] [CrossRef] [PubMed]
- Inekci, D.; Henriksen, K.; Linemann, T.; Karsdal, M.A.; Habib, A.; Bisgaard, C.; Eriksen, F.B.; Vilholm, O.J. Serum Fragments of Tau for the Differential Diagnosis of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Rinne, J.O.; Kadir, A.; Långström, B. The use of PET in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau protein and neurodegeneration. Semin. Cell Dev. Biol. 2004, 15, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Bigio, E.H.; Mackenzie, I.R.A.; Neumann, M.; Lee, V.M.-Y.; Hatanpaa, K.J.; White, C.L.; Schneider, J.A.; Grinberg, L.T.; Halliday, G.; et al. Consortium for Frontotemporal Lobar Degeneration Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Gambhir, S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Benzinger, T.L.; Su, Y.; Christensen, J.; Friedrichsen, K.; Aldea, P.; McConathy, J.; Cairns, N.J.; Fagan, A.M.; Morris, J.C.; Ances, B.M. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol. 2016, 73, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Brosch, J.R.; Farlow, M.R.; Risacher, S.L.; Apostolova, L.G. Tau Imaging in Alzheimer’s Disease Diagnosis and Clinical Trials. Neurotherapeutics 2017, 14, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Chien, D.T.; Bahri, S.; Szardenings, A.K.; Walsh, J.C.; Mu, F.; Su, M.-Y.; Shankle, W.R.; Elizarov, A.; Kolb, H.C. Early Clinical PET Imaging Results with the Novel PHF-Tau Radioligand [F18]-T807. J. Alzheimer’s Dis. 2013, 34, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.-F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.-R.; Trojanowski, J.Q.; Lee, V.M.-Y.; Ono, M.; et al. Imaging of Tau Pathology in a Tauopathy Mouse Model and in Alzheimer Patients Compared to Normal Controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Harada, R.; Yates, P.; Pejoska, S.; Kudo, Y.; Masters, C.L.; Yanai, K.; et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 2014, 137, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Marquié, M.; Normandin, M.D.; Vanderburg, C.R.; Costantino, I.M.; Bien, E.A.; Rycyna, L.G.; Klunk, W.E.; Mathis, C.A.; Ikonomovic, M.D.; Debnath, M.L.; et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann. Neurol. 2015, 78, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Schonhaut, D.R.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Ghosh, P.M.; Santos, M.; Miller, Z.A.; Bettcher, B.M.; Gorno-Tempini, M.L.; et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann. Neurol. 2015, 77, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Okamura, N. In vivo tau imaging: Obstacles and progress. Alzheimer’s Dement. 2014, 10, S254–S264. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Furumoto, S.; Fodero-Tavoletti, M.; Harada, R.; Mulligan, R.S.; Kudo, Y.; Masters, C.L.; Yanai, K.; Rowe, C.C.; Okamura, N. The challenges of tau imaging. Future Neurol. 2012, 7, 409–421. [Google Scholar] [CrossRef]
- Chien, D.T.; Szardenings, A.K.; Bahri, S.; Walsh, J.C.; Mu, F.; Xia, C.; Shankle, W.R.; Lerner, A.J.; Su, M.-Y.; Elizarov, A.; et al. Early Clinical PET Imaging Results with the Novel PHF-Tau Radioligand [F18]-T808. J. Alzheimer’s Dis. 2013, 38, 171–184. [Google Scholar] [CrossRef] [PubMed]
- James, O.G.; Doraiswamy, P.M.; Borges-Neto, S. PET Imaging of Tau Pathology in Alzheimer’s Disease and Tauopathies. Front. Neurol. 2015, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; Rabinovici, G.D.; Jagust, W.J. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Bakkour, A.; Salat, D.H.; Feczko, E.; Pacheco, J.; Greve, D.N.; Grodstein, F.; Wright, C.I.; Blacker, D.; Rosas, H.D.; et al. The Cortical Signature of Alzheimer’s Disease: Regionally Specific Cortical Thinning Relates to Symptom Severity in Very Mild to Mild AD Dementia and is Detectable in Asymptomatic Amyloid-Positive Individuals. Cereb. Cortex 2009, 19, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, P.; Whitwell, J.L.; Kantarci, K.; Josephs, K.A.; Parisi, J.E.; Shiung, M.S.; Knopman, D.S.; Boeve, B.F.; Petersen, R.C.; Dickson, D.W.; et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 2008, 42, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Benzinger, T.L.; Hassenstab, J.; Blazey, T.; Owen, C.; Liu, J.; Fagan, A.M.; Morris, J.C.; Ances, B.M. Spatially distinct atrophy is linked to -amyloid and tau in preclinical Alzheimer disease. Neurology 2015, 84, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Shimada, H.; Kitamura, S.; Shinotoh, H.; Endo, H.; Niwa, F.; Hirano, S.; Kimura, Y.; Zhang, M.-R.; Kuwabara, S.; Suhara, T.; et al. Association between Aβ and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: A [11C]PBB3-PET study. Alzheimer’s Dement. (Amsterdam, Netherlands) 2017, 6, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fodero-Tavoletti, M.T.; Okamura, N.; Furumoto, S.; Mulligan, R.S.; Connor, A.R.; McLean, C.A.; Cao, D.; Rigopoulos, A.; Cartwright, G.A.; O’Keefe, G.; et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011, 134, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Okamura, N.; Furumoto, S.; Tago, T.; Maruyama, M.; Higuchi, M.; Yoshikawa, T.; Arai, H.; Iwata, R.; Kudo, Y.; et al. Comparison of the binding characteristics of [18F]THK-523 and other amyloid imaging tracers to Alzheimer’s disease pathology. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Schofield, E.; Kersaitis, C.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: Tau-positive glia differentiate these disorders. Brain 2003, 126, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of Inflammatory Mediators and Microglial Activation in Mice Transgenic for Mutant Human P301S Tau Protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Cooper, J.D.; Hanger, D.P.; Noble, W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front. psychiatry 2010, 1, 136. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Garwood, C.; Stephenson, J.; Kinsey, A.M.; Hanger, D.P.; Anderton, B.H. Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. 2009, 23, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Borsini, A.; Zunszain, P.A.; Thuret, S.; Pariante, C.M. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015, 38, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.-W.; Ondrejcak, T.; Rowan, M.J. Glutamate receptors in preclinical research on Alzheimer’s disease: Update on recent advances. Pharmacol. Biochem. Behav. 2012, 100, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E. Alzheimer disease therapy: Can the amyloid cascade be halted? J. Clin. Invest. 2003, 111, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Paul, S.M.; DeMattos, R.B. Abeta immunization and anti-Abeta antibodies: Potential therapies for the prevention and treatment of Alzheimer’s disease. Adv. Drug Deliv. Rev. 2002, 54, 1603–1613. [Google Scholar] [CrossRef]
- Mikulca, J.A.; Nguyen, V.; Gajdosik, D.A.; Teklu, S.G.; Giunta, E.A.; Lessa, E.A.; Tran, C.H.; Terak, E.C.; Raffa, R.B. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J. Clin. Pharm. Ther. 2014, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Frisardi, V.; Solfrizzi, V.; Imbimbo, B.P.; Logroscino, G.; Santamato, A.; Greco, A.; Seripa, D.; Pilotto, A. Interacting with γ-secretase for treating Alzheimer’s disease: From inhibition to modulation. Curr. Med. Chem. 2011, 18, 5430–5447. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I. Tau as a Drug Target in Alzheimer’s Disease. J. Mol. Neurosci. 2002, 19, 337–338. [Google Scholar] [CrossRef]
- Loy, R.; Tariot, P.N. Neuroprotective properties of valproate: Potential benefit for AD and tauopathies. J. Mol. Neurosci. 2002, 19, 303–307. [Google Scholar] [CrossRef]
- Freland, L.; Beaulieu, J.-M. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 2012, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-M.; Xiong, Y.-S.; Kong, F.-L.; Qu, M.; Wang, Q.; Chen, X.-Q.; Wang, J.-Z.; Zhu, L.-Q. Neuroglobin attenuates Alzheimer-like tau hyperphosphorylation by activating Akt signaling. J. Neurochem. 2012, 120, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Tell, V.; Hilgeroth, A. Recent developments of protein kinase inhibitors as potential AD therapeutics. Front. Cell. Neurosci. 2013, 7, 189. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.T.; Sigurdsson, E.M. Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 2015, 21, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau–Tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers. Res. Ther. 2014, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Brunden, K.R.; Huryn, D.M.; Trojanowski, J.Q.; Lee, V.M.-Y.; Smith, A.B. III Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies. J. Med. Chem. 2012, 55, 8979–8996. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Nirzhor, S.; Akter, R. A Review of the Recent Advances Made with SIRT6 and its Implications on Aging Related Processes, Major Human Diseases, and Possible Therapeutic Targets. Biomolecules 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.S.; Choi, H.; Song, H.; Hwang, Y.J.; Kim, A.; Ryu, H.; Mook-Jung, I. p53-dependent SIRT6 expression protects Aβ42-induced DNA damage. Sci. Rep. 2016, 6, 25628. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Enzyme | Phosphorylation sites | Reference |
|---|---|---|
| PKA | Ser-195, Ser-198, Ser199, Ser-202, Ser-214, Ser-235, Ser-258, Ser-262, Ser-324, Ser-356, Ser-409, Ser-412, Ser-413, Ser422, Ser-435, Thr-205, Thr-212, Thr-217, Thr-231 | [45,46,47,48,49,50] |
| PKB/Akt | Ser-214, Thr-212 | [51] |
| PKC | Ser-258, Ser-293, Ser-324, Ser-352 | [52] |
| PKN | Ser-214, Ser-258, Ser-320, Ser-352 | [52] |
| AMPK | Ser-262, Ser-396, Ser-404, Thr-231 | [53,54] |
| CDK5 | Ser-199, Ser-202, Ser-214, Ser-235, Ser-396, Ser-404, Thr-181, Thr-205, Thr-212, Thr-217, Thr-231 | [55,56] |
| ERK 1/2 | Ser-46, Ser-199, Ser-202, Ser-235, Ser-396, Ser-404, Ser-422, Thr-50, Thr-153, Thr-181, Thr-205, Thr-212, Thr-217 | [57] |
| GSK-3β | Ser-46, Ser-184, Ser-199, Ser-202, Ser-214, Thr-50, Thr-181, Thr-205, Thr-212, Thr-217, Thr-231 | [56,58,59,60] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam Khan, R.; Nirzhor, S.S.R.; Rashid, B. A Closer Look into the Role of Protein Tau in the Identification of Promising Therapeutic Targets for Alzheimer’s Disease. Brain Sci. 2018, 8, 162. https://doi.org/10.3390/brainsci8090162
Islam Khan R, Nirzhor SSR, Rashid B. A Closer Look into the Role of Protein Tau in the Identification of Promising Therapeutic Targets for Alzheimer’s Disease. Brain Sciences. 2018; 8(9):162. https://doi.org/10.3390/brainsci8090162
Chicago/Turabian StyleIslam Khan, Rubayat, Saif Shahriar Rahman Nirzhor, and Barnaly Rashid. 2018. "A Closer Look into the Role of Protein Tau in the Identification of Promising Therapeutic Targets for Alzheimer’s Disease" Brain Sciences 8, no. 9: 162. https://doi.org/10.3390/brainsci8090162
APA StyleIslam Khan, R., Nirzhor, S. S. R., & Rashid, B. (2018). A Closer Look into the Role of Protein Tau in the Identification of Promising Therapeutic Targets for Alzheimer’s Disease. Brain Sciences, 8(9), 162. https://doi.org/10.3390/brainsci8090162