
Multiple Sclerosis: Immunopathology and Treatment Update


 ,
, 

Abstract

1. Introduction
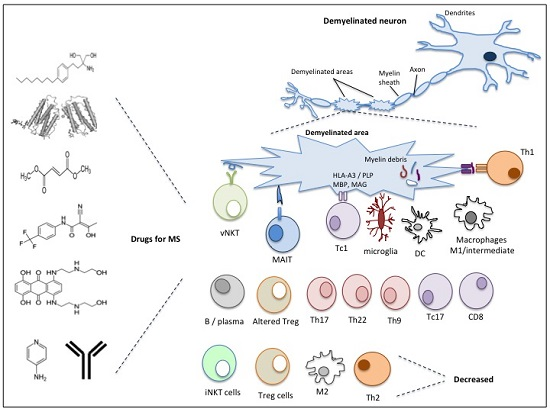
2. Immunopathophysiology of MS
2.1. Natural Killer T (NKT) Cells
2.2. Mucosal-Associated Invariant T (MAIT) Cells
2.3. Regulatory T Cells (Tregs)
2.4. Macrophages and Microglia
2.5. T Helper Cells
2.6. CD8 T Cells
2.7. B Cells
2.8. Dendritic Cells
2.9. Myeloid Derived Suppressor Cells
3. Current Drug Therapies for Multiple Sclerosis
3.1. Treatment of MS Relapses
3.2. Long-Term Treatment of MS with Disease-Modifying Agents
3.2.1. Interferons (Avonex®, Biogen, Cambridge, MA, USA; Betaseron®, Bayer, Leverkusen, Germany; Extavia®, Novartis Pharma AG, Basel, Switzerland; Rebif®, EMD Serono Inc., Darmstadt, Germany; Plegridy®, Biogen, Cambridge, MA, USA)
3.2.2. Glatiramer Acetate (Copaxone®, Inc., Petah Tikva, Israel)
3.2.3. Dimethyl Fumarate (Tecfidera®, Biogen, Cambridge, MA, USA)
3.2.4. Teriflunomide (Aubagio®, Sanofi Genzyme, Cambridge, MA, USA)
3.2.5. Fingolimod (Gilenya®, FTY720, Novartis Pharma AG, Basel, Switzerland)
3.2.6. Mitoxantrone (Novantrone®, Immunex/Amgen, Thousand Oaks, CA, USA)
3.3. Treatment Using Humanized Monoclonal Antibodies
3.3.1. Natalizumab (Tysabr®, Biogen, Cambridge, MA, USA)
3.3.2. Ofatumumab (Arzerra®, Novartis Pharma AG, Basel, Switzerland)
3.3.3. Ocrelizumab (Ocrevus®, Genentech Inc., San Fransisco, CA, USA)
3.3.4. Alemtuzumab (Lemtrada®, Sanofi Genzyme, Cambridge, MA, USA)
3.3.5. Daclizumab (Zinbryta®, Biogen, Cambridge, MA, USA)
4. New and Emerging Immunotherapeutic Strategies against MS
4.1. Stem Cells
4.2. DNA Vaccine Studies
4.3. Nanoparticles
4.4. Altered Peptide Ligands
4.4.1. Cyclic Peptides
4.4.2. Mannan as a Carrier to Modulate Immune Responses
5. Symptomatic Medication
Dalfampridine (Ampyra/Fampyra®, Acorda Therapeutics)
6. Conclusions and Future Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Grytten, N.; Torkildsen, O.; Myhr, K.M. Time trends in the incidence and prevalence of multiple sclerosis in norway during eight decades. Acta Neurol. Scand. 2015, 132, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.; Antel, S.; Caramanos, Z.; Arnold, D.L.; Kuhlmann, T. Primary progressive multiple sclerosis: Part of the ms disease spectrum or separate disease entity? Acta Neuropathol. 2012, 123, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Sadovnick, A.D.; Ebers, G.C.; Dyment, D.A.; Risch, N.J. Evidence for genetic basis of multiple sclerosis. Lancet 1996, 347, 1728–1730. [Google Scholar] [CrossRef]
- Dai, H.; Ciric, B.; Zhang, G.X.; Rostami, A. Interleukin-10 plays a crucial role in suppression of experimental autoimmune encephalomyelitis by bowman-birk inhibitor. J. Neuroimmunol. 2012, 245, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Nessler, S.; Zhou, D.; Kieseier, B.; Hartung, H.P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, P. Improving ms patient care. J. Neurol. Suppl. 2004, 251, v69–v73. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Matsoukas, J.; Deraos, G.; Apostolopoulos, V. Towards immunotherapeutic drugs and vaccines against multiple sclerosis. Acta Biochim. Biophys. Sin. 2008, 40, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, C.; Bhatti, M.T. Currently approved and emerging oral therapies in multiple sclerosis: An update for the ophthalmologist. Surv. Ophthalmol. 2016, 61, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the mcdonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Lunde Larsen, L.S.; Larsson, H.B.W.; Frederiksen, J.L. The value of conventional high-field mri in ms in the light of the mcdonald criteria: A literature review. Acta Neurol. Scand. 2010, 122, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Gafson, A.; Giovannoni, G.; Hawkes, C.H. The diagnostic criteria for multiple sclerosis: From charcot to mcdonald. Mult. Scler. Relat. Disord. 2012, 1, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef]
- Bennett, J.; Basivireddy, J.; Kollar, A.; Biron, K.E.; Reickmann, P.; Jefferies, W.A.; McQuaid, S. Blood–brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model eae. J. Neuroimmunol. 2010, 229, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Farjam, M.; Zhang, G.X.; Ciric, B.; Rostami, A. Emerging immunopharmacological targets in multiple sclerosis. J. Neurol. Sci. 2015, 358, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.A.; Wu, G.F.; Pewe, L.; Perlman, S. Axonal damage is t cell mediated and occurs concomitantly with demyelination in mice infected with a neurotropic coronavirus. J. Virol. 2001, 75, 6115–6120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Kelly, K.A. Phenotype and function of regulatory t cells in the genital tract. Curr. Trends Immunol. 2011, 12, 89–94. [Google Scholar] [PubMed]
- Bianchini, E.; De Biasi, S.; Simone, A.M.; Ferraro, D.; Sola, P.; Cossarizza, A.; Pinti, M. Invariant natural killer T cells and mucosal-associated invariant T cells in multiple sclerosis. Immunol. Lett. 2017, 183, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tabarkiewicz, J.; Pogoda, K.; Karczmarczyk, A.; Pozarowski, P.; Giannopoulos, K. The role of il-17 and th17 lymphocytes in autoimmune diseases. Arch. Immunol. Ther. Exp. 2015, 63, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Van Hamburg, J.P.; Asmawidjaja, P.S.; Davelaar, N.; Mus, A.M.C.; Colin, E.M.; Hazes, J.M.W.; Dolhain, R.J.E.M.; Lubberts, E. Th17 cells, but not th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17a production. Arthritis Rheum. 2011, 63, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Dolati, S.; Babaloo, Z.; Jadidi-Niaragh, F.; Ayromlou, H.; Sadreddini, S.; Yousefi, M. Multiple sclerosis: Therapeutic applications of advancing drug delivery systems. Biomed. Pharmacother. 2017, 86, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Münzel, E.J.; Williams, A. Promoting remyelination in multiple sclerosis-recent advances. Drugs 2013, 73, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Inglese, M.; Petracca, M. Therapeutic strategies in multiple sclerosis: A focus on neuroprotection and repair and relevance to schizophrenia. Schizophr. Res. 2015, 161, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Koriem, K.M.M. Multiple sclerosis: New insights and trends. Asian Pac. J. Trop. Biomed. 2016, 6, 429–440. [Google Scholar] [CrossRef]
- Kallaur, A.P.; Lopes, J.; Oliveira, S.R.; Simão, A.N.; Reiche, E.M.; de Almeida, E.R.D.; Morimoto, H.K.; de Pereira, W.L.; Alfieri, D.F.; Borelli, S.D.; et al. Immune-inflammatory and oxidative and nitrosative stress biomarkers of depression symptoms in subjects with multiple sclerosis: Increased peripheral inflammation but less acute neuroinflammation. Mol. Neurobiol. 2016, 53, 5191–5202. [Google Scholar] [CrossRef] [PubMed]
- Mirshafiey, A.; Jadidi-Niaragh, F. Prostaglandins in pathogenesis and treatment of multiple sclerosis. Immunopharmacol. Immunotoxicol. 2010, 32, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; Van Horssen, J.; Lassmann, H. Nadph oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Wu, L.; Parekh, V.V. Natural killer t cells in multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. Immunology 2015, 146, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gigli, G.; Caielli, S.; Cutuli, D.; Falcone, M. Innate immunity modulates autoimmunity: Type 1 interferon-beta treatment in multiple sclerosis promotes growth and function of regulatory invariant natural killer t cells through dendritic cell maturation. Immunology 2007, 122, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Kondo, T.; Gumperz, J.E.; Brenner, M.B.; Miyake, S.; Yamamura, T. Th2 bias of cd4+ nkt cells derived from multiple sclerosis in remission. Int. Immunol. 2003, 15, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Mars, L.T.; Laloux, V.; Goude, K.; Desbois, S.; Saoudi, A.; Van Kaer, L.; Lassmann, H.; Herbelin, A.; Lehuen, A.; Liblau, R.S. Cutting edge: V alpha 14-j alpha 281 nkt cells naturally regulate experimental autoimmune encephalomyelitis in nonobese diabetic mice. J. Immunol. 2002, 168, 6007–6011. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L. Alpha-galactosylceramide therapy for autoimmune diseases: Prospects and obstacles. Nat. Rev. Immunol. 2005, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant nk t cells: Potential for immunotherapeutic targeting with glycolipid antigens. Immunotherapy 2011, 3, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Jahng, A.; Maricic, I.; Aguilera, C.; Cardell, S.; Halder, R.C.; Kumar, V. Prevention of autoimmunity by targeting a distinct, noninvariant cd1d-reactive t cell population reactive to sulfatide. J. Exp. Med. 2004, 199, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Napier, R.J.; Adams, E.J.; Gold, M.C.; Lewinsohn, D.M. The role of mucosal associated invariant t cells in antimicrobial immunity. Front. Immunol. 2015, 6, 344. [Google Scholar] [CrossRef] [PubMed]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. Mr1 presents microbial vitamin b metabolites to mait cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, S.V.; Angelini, D.F.; Dubinsky, A.N.; Morel, E.; Oh, U.; Jones, J.L.; Carassiti, D.; Reynolds, R.; Salvetti, M.; Calabresi, P.A.; et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes il-17 producing mucosal-associated invariant t cells in multiple sclerosis. Brain 2013, 136, 2888–2903. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Miyake, S.; Chiba, A.; Lantz, O.; Yamamura, T. Mucosal-associated invariant t cells regulate th1 response in multiple sclerosis. Int. Immunol. 2011, 23, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory t cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Koldzic, D.N.; Izikson, L.; Reddy, J.; Nazareno, R.F.; Sakaguchi, S.; Kuchroo, V.K.; Weiner, H.L. Il-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory t cells. Int. Immunol. 2004, 16, 249–256. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Stephens, L.A.; Anderton, S.M. Natural recovery and protection from autoimmune encephalomyelitis: Contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 2005, 175, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Bakke, A.C.; Hopke, C.; Dwyer, J.; Vandenbark, A.A.; Offner, H. Estrogen treatment induces a novel population of regulatory cells, which suppresses experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2004, 77, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Prod’homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type ii monocytes modulate t cell-mediated central nervous system autoimmune disease. Nat. Med. 2007, 13, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Beyersdorf, N.; Gaupp, S.; Balbach, K.; Schmidt, J.; Toyka, K.V.; Lin, C.H.; Hanke, T.; Hunig, T.; Kerkau, T.; Gold, R. Selective targeting of regulatory t cells with cd28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J. Exp. Med. 2005, 202, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.L.; Wiendl, H. The role of regulatory t cells in multiple sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Diebold, M.; Derfuss, T. Immunological treatment of multiple sclerosis. Semin. Hematol. 2016, 53 (Suppl. 1), S54–S57. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Sommermeier, N.; Bergmann, M.; Zettl, U.; Goebel, H.H.; Kretzschmar, H.A.; Lassmann, H. Macrophages in multiple sclerosis. Immunobiology 1996, 195, 588–600. [Google Scholar] [CrossRef]
- Vogel, D.Y.; Vereyken, E.J.; Glim, J.E.; Heijnen, P.D.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Peferoen, L.A.; Vogel, D.Y.; Ummenthum, K.; Breur, M.; Heijnen, P.D.; Gerritsen, W.H.; Peferoen-Baert, R.M.; van der Valk, P.; Dijkstra, C.D.; Amor, S. Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during cns remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Yu, J.; Feng, L.; Hou, S.; Liu, Y.; Guo, M.; Xie, Y.; Meng, J.; Zhang, H.; et al. Targeting the shift from m1 to m2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS ONE 2013, 8, e54841. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; de Courten, M.P.; Stojanovska, L.; Blatch, G.L.; Tangalakis, K.; de Courten, B. The complex immunological and inflammatory network of adipose tissue in obesity. Mol. Nutr. Food Res. 2016, 60, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, Q.; Mao, G.; Dowling, C.A.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl fumarate selectively reduces memory t cells and shifts the balance between th1/th17 and th2 in multiple sclerosis patients. J. Immunol. 2017, 198, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Deraos, G.; Matsoukas, M.T.; Day, S.; Stojanovska, L.; Tselios, T.; Androutsou, M.E.; Matsoukas, J. Cyclic citrullinated mbp87–99 peptide stimulates t cell responses: Implications in triggering disease. Bioorg. Med. Chem. 2017, 25, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Deraos, G.; Chatzantoni, K.; Matsoukas, M.T.; Tselios, T.; Deraos, S.; Katsara, M.; Papathanasopoulos, P.; Vynios, D.; Apostolopoulos, V.; Mouzaki, A.; et al. Citrullination of linear and cyclic altered peptide ligands from myelin basic protein (mbp(87–99)) epitope elicits a th1 polarized response by t cells isolated from multiple sclerosis patients: Implications in triggering disease. J. Med. Chem. 2008, 51, 7834–7842. [Google Scholar] [CrossRef] [PubMed]
- Deraos, G.; Rodi, M.; Kalbacher, H.; Chatzantoni, K.; Karagiannis, F.; Synodinos, L.; Plotas, P.; Papalois, A.; Dimisianos, N.; Papathanasopoulos, P.; et al. Properties of myelin altered peptide ligand cyclo(87–99)(ala91,ala96)mbp87–99 render it a promising drug lead for immunotherapy of multiple sclerosis. Eur. J. Med. Chem. 2015, 101, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Deraos, G.; Tselios, T.; Matsoukas, J.; Apostolopoulos, V. Design of novel cyclic altered peptide ligands of myelin basic protein mbp83–99 that modulate immune responses in sjl/j mice. J. Med. Chem. 2008, 51, 3971–3978. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Deraos, G.; Tselios, T.; Matsoukas, M.T.; Friligou, I.; Matsoukas, J.; Apostolopoulos, V. Design and synthesis of a cyclic double mutant peptide (cyclo(87–99)[a91,a96]mbp87–99) induces altered responses in mice after conjugation to mannan: Implications in the immunotherapy of multiple sclerosis. J. Med. Chem. 2009, 52, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Deraos, S.; Tselios, T.V.; Pietersz, G.; Matsoukas, J.; Apostolopoulos, V. Immune responses of linear and cyclic plp139–151 mutant peptides in sjl/j mice: Peptides in their free state versus mannan conjugation. Immunotherapy 2014, 6, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Minigo, G.; Plebanski, M.; Apostolopoulos, V. The good, the bad and the ugly: How altered peptide ligands modulate immunity. Expert Opin. Biol. Ther. 2008, 8, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Tselios, T.; Deraos, S.; Deraos, G.; Matsoukas, M.T.; Lazoura, E.; Matsoukas, J.; Apostolopoulos, V. Round and round we go: Cyclic peptides in disease. Curr. Med. Chem. 2006, 13, 2221–2232. [Google Scholar] [PubMed]
- Katsara, M.; Yuriev, E.; Ramsland, P.A.; Deraos, G.; Tselios, T.; Matsoukas, J.; Apostolopoulos, V. A double mutation of mbp83–99 peptide induces il-4 responses and antagonizes ifn-γ responses. J. Neuroimmunol. 2008, 200, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Yuriev, E.; Ramsland, P.A.; Deraos, G.; Tselios, T.; Matsoukas, J.; Apostolopoulos, V. Mannosylation of mutated mbp83–99 peptides diverts immune responses from th1 to th2. Mol. Immunol. 2008, 45, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Yuriev, E.; Ramsland, P.A.; Tselios, T.; Deraos, G.; Lourbopoulos, A.; Grigoriadis, N.; Matsoukas, J.; Apostolopoulos, V. Altered peptide ligands of myelin basic protein (mbp87–99) conjugated to reduced mannan modulate immune responses in mice. Immunology 2009, 128, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Apostolopoulos, V.; Kalbacher, H.; Papini, A.M.; Tselios, T.; Chatzantoni, K.; Biagioli, T.; Lolli, F.; Deraos, S.; Papathanassopoulos, P.; et al. Design and synthesis of a novel potent myelin basic protein epitope 87–99 cyclic analogue: Enhanced stability and biological properties of mimics render them a potentially new class of immunomodulators. J. Med. Chem. 2005, 48, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Tselios, T.; Apostolopoulos, V.; Daliani, I.; Deraos, S.; Grdadolnik, S.; Mavromoustakos, T.; Melachrinou, M.; Thymianou, S.; Probert, L.; Mouzaki, A.; et al. Antagonistic effects of human cyclic mbp(87–99) altered peptide ligands in experimental allergic encephalomyelitis and human t-cell proliferation. J. Med. Chem. 2002, 45, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Tselios, T.V.; Lamari, F.N.; Karathanasopoulou, I.; Katsara, M.; Apostolopoulos, V.; Pietersz, G.A.; Matsoukas, J.M.; Karamanos, N.K. Synthesis and study of the electrophoretic behavior of mannan conjugates with cyclic peptide analogue of myelin basic protein using lysine-glycine linker. Anal. Biochem. 2005, 347, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tseveleki, V.; Tselios, T.; Kanistras, I.; Koutsoni, O.; Karamita, M.; Vamvakas, S.S.; Apostolopoulos, V.; Dotsika, E.; Matsoukas, J.; Lassmann, H.; et al. Mannan-conjugated myelin peptides prime non-pathogenic th1 and th17 cells and ameliorate experimental autoimmune encephalomyelitis. Exp. Neurol. 2015, 267, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.; Chang, C.; Lu, L.; Lau, C.S.; Lu, Q. Th9 cells and il-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum. Immunol. 2017, 78, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Batistini, L.; Borsellino, G. Advances in t helper 17 cell biology: Pathogenic role and potential therapy in multiple sclerosis. Mediat. Inflamm. 2015, 475158. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; Bardina, V.; De Mercanti, S.; Quaglino, P.; De Palma, R.; Gned, D.; Brusa, D.; Durelli, L.; Novelli, F.; Clerico, M. Th22 cells are expanded in multiple sclerosis and are resistant to ifn-beta. J. Leukoc. Biol. 2014, 96, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Muls, N.; Nasr, Z.; Dang, H.A.; Sindic, C.; van Pesch, V. Il-22, gm-csf and il-17 in peripheral CD4+ t cell subpopulations during multiple sclerosis relapses and remission. Impact of corticosteroid therapy. PLoS ONE 2017, 12, e0173780. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Fujio, K.; Okamura, T.; Yamamoto, K. Interleukin-27 in t cell immunity. Int. J. Mol. Sci. 2015, 16, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Naderi, S.; Hejazi, Z.; Shajarian, M.; Alsahebfosoul, F.; Etemadifar, M.; Sedaghat, N. Il-27 plasma level in relapsing remitting multiple sclerosis subjects: The double-faced cytokine. J. Immunoass. Immunochem. 2016, 37, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Senecal, V.; Deblois, G.; Beauseigle, D.; Schneider, R.; Brandenburg, J.; Newcombe, J.; Moore, C.S.; Prat, A.; Antel, J.; Arbour, N. Production of il-27 in multiple sclerosis lesions by astrocytes and myeloid cells: Modulation of local immune responses. Glia 2016, 64, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Kawanokuchi, J.; Takeuchi, H.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Interleukin-27 promotes inflammatory and neuroprotective responses in microglia. Clin. Exp. Neuroimmunol. 2013, 4, 36–45. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G. The major histocompatibility complex and multiple sclerosis: A smoking gun? Brain 2011, 134, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Dressel, A.; Chin, J.L.; Sette, A.; Gausling, R.; Hollsberg, P.; Hafler, D.A. Autoantigen recognition by human cd8 t cell clones: Enhanced agonist response induced by altered peptide ligands. J. Immunol. 1997, 159, 4943–4951. [Google Scholar] [PubMed]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating t cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Salehi, Z.; Doosti, R.; Beheshti, M.; Janzamin, E.; Sahraian, M.A.; Izad, M. Differential frequency of CD8+ T cell subsets in multiple sclerosis patients with various clinical patterns. PLoS ONE 2016, 11, e0159565. [Google Scholar] [CrossRef] [PubMed]
- Disanto, G.; Morahan, J.M.; Barnett, M.H.; Giovannoni, G.; Ramagopalan, S.V. The evidence for a role of b cells in multiple sclerosis. Neurology 2012, 78, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Wekerle, H. B cells in multiple sclerosis. Autoimmunity 2017, 50, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Winger, R.C.; Zamvil, S.S. Antibodies in multiple sclerosis oligoclonal bands target debris. Proc. Natl. Acad. Sci. USA 2016, 113, 7696–7698. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.M.; Xiao, B.G.; Ozenci, V.; Kouwenhoven, M.; Teleshova, N.; Fredrikson, S.; Link, H. Multiple sclerosis is associated with high levels of circulating dendritic cells secreting pro-inflammatory cytokines. J. Neuroimmunol. 1999, 99, 82–90. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Fuchsberger, M.; Xiang, S.D.; Apostolopoulos, V.; Plebanski, M. Myeloid derived suppressor cells and their role in diseases. Curr. Med. Chem. 2013, 20, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Wegner, A.; Verhagen, J.; Wraith, D.C. Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology 2017, 151, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through ido expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Filippini, G.; Brusaferri, F.; Sibley, W.A.; Citterio, A.; Ciucci, G.; Midgard, R.; Candelise, L. Corticosteroids or acth for acute exacerbations in multiple sclerosis. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Havrdova, E.; Zivadinov, R.; Krasensky, J.; Dwyer, M.G.; Novakova, I.; Dolezal, O.; Ticha, V.; Dusek, L.; Houzvickova, E.; Cox, J.L.; et al. Randomized study of interferon beta-1a, low-dose azathioprine, and low-dose corticosteroids in multiple sclerosis. Mult. Scler. 2009, 15, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Morrow, S.A.; Metz, L.M.; Kremenchutzky, M. High dose oral steroids commonly used to treat relapses in canadian ms clinics. Can. J. Neurol. Sci. 2009, 36, 213–215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Myhr, K.M.; Mellgren, S.I. Corticosteroids in the treatment of multiple sclerosis. Acta Neurol. Scand. 2009, 120, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Van Der Voort, L.F.; Visser, A.; Knol, D.L.; Oudejans, C.B.M.; Polman, C.H.; Killestein, J. Lack of interferon-beta bioactivity is associated with the occurrence of relapses in multiple sclerosis. Eur. J. Neurol. 2009, 16, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Freedman, M.S.; Polman, C.H.; Edan, G.; Hartung, H.-P.; Miller, D.H.; Montalbán, X.; Barkhof, F.; Radü, E.-W.; Bauer, L.; et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: A 3-year follow-up analysis of the benefit study. Lancet 2007, 370, 389–397. [Google Scholar] [CrossRef]
- Kappos, L.; Polman, C.H.; Freedman, M.S.; Edan, G.; Hartung, H.P.; Miller, D.H.; Montalban, X.; Barkhof, F.; Bauer, L.; Jakobs, P.; et al. Treatment with interferon beta-1b delays conversion to clinically definite and mcdonald ms in patients with clinically isolated syndromes. Neurology 2006, 67, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.R. Challenges in randomized controlled trials and emerging multiple sclerosis therapeutics. Neurosci. Bull. 2015, 31, 745–754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fenu, G.; Lorefice, L.; Frau, F.; Coghe, G.C.; Marrosu, M.G.; Cocco, E. Induction and escalation therapies in multiple sclerosis. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2015, 14, 26–34. [Google Scholar] [CrossRef]
- Kipp, M.; Wagenknecht, N.; Beyer, C.; Samer, S.; Wuerfel, J.; Nikoubashman, O. Thalamus pathology in multiple sclerosis: From biology to clinical application. Cell. Mol. Life Sci. 2015, 72, 1127–1147. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Radue, E.W.; Goodin, D.; Jeffery, D.; Rammohan, K.W.; Reder, A.T.; Vollmer, T.; Agius, M.A.; Kappos, L.; Stites, T.; et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (freedoms ii): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 545–556. [Google Scholar] [CrossRef]
- Greenberg, B.M.; Balcer, L.; Calabresi, P.A.; Cree, B.; Cross, A.; Frohman, T.; Gold, R.; Havrdova, E.; Hemmer, B.; Kieseier, B.C.; et al. Interferon beta use and disability prevention in relapsing-remitting multiple sclerosis. JAMA Neurol. 2013, 70, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Noyes, K.; Weinstock-Guttman, B. Impact of diagnosis and early treatment on the course of multiple sclerosis. Am. J. Manag. Care 2013, 19, s321–s331. [Google Scholar]
- Shirani, A.; Zhao, Y.; Karim, M.E.; Evans, C.; Kingwell, E.; Van Der Kop, M.L.; Oger, J.; Gustafson, P.; Petkau, J.; Tremlett, H. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA 2012, 308, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Stüve, O. Management of secondary progressive multiple sclerosis: Prophylactic treatment - past, present, and future aspects. Curr. Treat. Options Neurol. 2013, 15, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Trojano, M.; Pellegrini, F.; Paolicelli, D.; Fuiani, A.; Zimatore, G.B.; Tortorella, C.; Simone, I.L.; Patti, F.; Ghezzi, A.; Zipoli, V.; et al. Real-life impact of early interferonβ therapy in relapsing multiple sclerosis. Ann. Neurol. 2009, 66, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Goodin, D.S.; Ebers, G.C.; Cutter, G.; Cook, S.D.; O'Donnell, T.; Reder, A.T.; Kremenchutzky, M.; Oger, J.; Rametta, M.; Beckmann, K.; et al. Cause of death in ms: Long-term follow-up of a randomised cohort, 21 years after the start of the pivotal ifnβ-1b study. BMJ Open 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Goodin, D.S.; Reder, A.T.; Ebers, G.C.; Cutter, G.; Kremenchutzky, M.; Oger, J.; Langdon, D.; Rametta, M.; Beckmann, K.; DeSimone, T.M.; et al. Survival in ms a randomized cohort study 21 years after the start of the pivotal ifnβ-1b trial. Neurology 2012, 78, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Mitsdoerffer, M.; Kuchroo, V. New pieces in the puzzle: How does interferon-beta really work in multiple sclerosis? Ann. Neurol. 2009, 65, 487–488. [Google Scholar] [CrossRef] [PubMed]
- The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 1993, 43, 655–661. [Google Scholar]
- The IFNB Multiple Sclerosis Study Group; The University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: Final outcome of the randomized controlled trial. Neurology 1995, 45, 1277–1285. [Google Scholar]
- Yong, V.W.; Giuliani, F.; Xue, M.; Bar-Or, A.; Metz, L.M. Experimental models of neuroprotection relevant to multiple sclerosis. Neurology 2007, 68, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Narayana, P.A.; O’Connor, P.; Coyle, P.K.; Ford, C.; Johnson, K.; Miller, A.; Pardo, L.; Kadosh, S.; Ladkani, D.; et al. Glatiramer acetate in primary progressive multiple sclerosis: Results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann. Neurol. 2007, 61, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S. Copolymer 1: A most reasonable alternative therapy for early relapsing–remitting multiple sclerosis with mild disability. Neurology 1995, 45, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Farina, C.; Wekerle, H.; Hohlfeld, R. Mechanisms of action of glatiramer acetate in multiple sclerosis. Neurology 2001, 56, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Ragheb, S.; Abramczyk, S.; Lisak, D.; Lisak, R. Long-term therapy with glatiramer acetate in multiple sclerosis: Effect on t-cells. Mult. Scler. 2001, 7, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Korporal, M.; Balint, B.; Fritzsching, B.; Schwarz, A.; Wildemann, B. Glatiramer acetate improves regulatory t-cell function by expansion of naive CD4(+)CD25(+)Foxp3(+)CD31(+) t-cells in patients with multiple sclerosis. J. Neuroimmunol. 2009, 216, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B.; et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase iii multicenter, double-blind placebo-controlled trial. Neurology 1995, 45, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Moharregh-Khiabani, D.; Linker, R.A.; Gold, R.; Stangel, M. Fumaric acid and its esters: An emerging treatment for multiple sclerosis. Curr. Neuropharmacol. 2009, 7, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, P.; Bouchachia, I.; Goebels, N.; Henke, N.; Hofstetter, H.H.; Issberner, A.; Kovacs, Z.; Lewerenz, J.; Lisak, D.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. Teriflunomide, an inhibitor of dihydroorotate dehydrogenase for the potential oral treatment of multiple sclerosis. Curr. Opin. Investig. Drugs 2010, 11, 1313–1323. [Google Scholar] [PubMed]
- Korn, T.; Magnus, T.; Toyka, K.; Jung, S. Modulation of effector cell functions in experimental autoimmune encephalomyelitis by leflunomide--mechanisms independent of pyrimidine depletion. J. Leukoc. Biol. 2004, 76, 950–960. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.; Wolinsky, J.S.; Confavreux, C.; Comi, G.; Kappos, L.; Olsson, T.P.; Benzerdjeb, H.; Truffinet, P.; Wang, L.; Miller, A.; et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med. 2011, 365, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Sanvito, L.; Constantinescu, C.S.; Gran, B. Novel therapeutic approaches to autoimmune demyelinating disorders. Curr. Pharm. Des. 2011, 17, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.A. Current therapeutic options in pediatric multiple sclerosis. Curr. Treat. Options Neurol. 2011, 13, 544–559. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.W.; Li, D.; Freedman, M.S.; Bar-Or, A.; Rice, G.P.A.; Confavreux, C.; Paty, D.W.; Stewart, J.A.; Scheyer, R. A phase ii study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology 2006, 66, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; O’Connor, P.; Comi, G.; Freedman, M.S.; Miller, A.E.; Olsson, T.P.; Wolinsky, J.S.; Bagulho, T.; Delhay, J.-L.; Dukovic, D.; et al. Oral teriflunomide for patients with relapsing multiple sclerosis (tower): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 247–256. [Google Scholar] [CrossRef]
- Vermersch, P.; Czlonkowska, A.; Grimaldi, L.M.; Confavreux, C.; Comi, G.; Kappos, L.; Olsson, T.P.; Benamor, M.; Bauer, D.; Truffinet, P.; et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: A randomised, controlled phase 3 trial. Mult. Scler. J. 2014, 20, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator fty720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on s1p receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. Fty720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (s1p1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, Q.T.; Song, S.S.; Wu, Y.J.; Ma, Y.K.; Zhang, L.L.; Chen, J.Y.; Wu, H.X.; Jiang, L.; Wei, W. Combined use of etanercept and mtx restores CD4+/CD8+ ratio and tregs in spleen and thymus in collagen-induced arthritis. Inflamm. Res. 2012, 61, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Lenk, H.; Muller, U.; Tanneberger, S. Mitoxantrone: Mechanism of action, antitumor activity, pharmacokinetics, efficacy in the treatment of solid tumors and lymphomas, and toxicity. Anticancer Res. 1987, 7, 1257–1264. [Google Scholar] [PubMed]
- Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [Google Scholar] [CrossRef]
- Edan, G.; Miller, D.; Clanet, M.; Confavreux, C.; Lyon-Caen, O.; Lubetzki, C.; Brochet, B.; Berry, I.; Rolland, Y.; Froment, J.C.; et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: A randomised multicentre study of active disease using mri and clinical criteria. J. Neurol. Neurosurg. Psychiatry 1997, 62, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, V.; Radaelli, M.; Straffi, L.; Rodegher, M.; Comi, G. Mitoxantrone: Benefits and risks in multiple sclerosis patients. Neurol. Sci. 2009, 30, S167–S170. [Google Scholar] [CrossRef] [PubMed]
- Sheremata, W.A.; Minagar, A.; Alexander, J.S.; Vollmer, T. The role of alpha-4 integrin in the aetiology of multiple sclerosis: Current knowledge and therapeutic implications. CNS Drugs 2005, 19, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.P.A.; Hartung, H.P.; Calabresi, P.A. Anti-α4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 2005, 64, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Gold, R.; Hemmer, B.; Korn, T.; Zipp, F.; Hohlfeld, R.; Kieseier, B.C.; Wiendl, H. Diagnosis of multiple sclerosis 2010 revision of the mcdonald criteria. Nervenarzt 2011, 82, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Hohlfeld, R.; Voltz, R. Diagnosis and therapy of multiple sclerosis—Update 2003. MMW Fortschr. Med. 2003, 145, 88–95. [Google Scholar] [PubMed]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.A.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Natalizumab: New drug. Multiple sclerosis: Risky market approval. Prescrire Int. 2008, 17, 7–10.
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis--the plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lisby, S.; Grove, R.; Derosier, F.; Shackelford, S.; Havrdova, E.; Drulovic, J.; Filippi, M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: A phase 2 study. Neurology 2014, 82, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, W.K.; Munk, M.E.; Mackus, W.J.; van den Brakel, J.H.; Pluyter, M.; Glennie, M.J.; van de Winkel, J.G.; Parren, P.W. Estimation of dose requirements for sustained in vivo activity of a therapeutic human anti-cd20 antibody. Br. J. Haematol. 2008, 140, 303–312. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Identifier: NCT02792218 and NCT02792231. Available online: https://www.mdanderson.org/patients-family/diagnosis-treatment/clinical-trials.html (accessed on 20 June 2017).
- U.S. Food & Drug Administration (FDA). Drugs@fda: FDA Approved Drug Products. Available online: http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=761053 (accessed on 20 June 2017).
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Lycke, J. Monoclonal antibody therapies for the treatment of relapsing-remitting multiple sclerosis: Differentiating mechanisms and clinical outcomes. Ther. Adv. Neurol. Disord. 2015, 8, 274–293. [Google Scholar] [CrossRef] [PubMed]
- McAllister, L.D.; Beatty, P.G.; Rose, J. Allogeneic bone marrow transplant for chronic myelogenous leukemia in a patient with multiple sclerosis. Bone Marrow Transplant. 1997, 19, 395–397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mancardi, G.; Saccardi, R. Autologous haematopoietic stem-cell transplantation in multiple sclerosis. Lancet Neurol. 2008, 7, 626–636. [Google Scholar] [CrossRef]
- Sormani, M.P.; Muraro, P.A.; Schiavetti, I.; Signori, A.; Laroni, A.; Saccardi, R.; Mancardi, G.L. Autologous hematopoietic stem cell transplantation in multiple sclerosis: A meta-analysis. Neurology 2017, 88, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Yamout, B.; Hourani, R.; Salti, H.; Barada, W.; El-Hajj, T.; Al-Kutoubi, A.; Herlopian, A.; Baz, E.K.; Mahfouz, R.; Khalil-Hamdan, R.; et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J. Neuroimmunol. 2010, 227, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Llufriu, S.; Sepulveda, M.; Blanco, Y.; Marin, P.; Moreno, B.; Berenguer, J.; Gabilondo, I.; Martinez-Heras, E.; Sola-Valls, N.; Arnaiz, J.A.; et al. Randomized placebo-controlled phase ii trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE 2014, 9, e113936. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Fiol, M. Bht-3009, a myelin basic protein-encoding plasmid for the treatment of multiple sclerosis. Curr. Opin. Mol. Ther. 2009, 11, 463–470. [Google Scholar] [PubMed]
- Kang, Y.; Sun, Y.; Zhang, J.; Gao, W.; Kang, J.; Wang, Y.; Wang, B.; Xia, G. Treg cell resistance to apoptosis in DNA vaccination for experimental autoimmune encephalomyelitis treatment. PLoS ONE 2012, 7, e49994. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Vollmer, T.; Antel, J.; Arnold, D.L.; Bodner, C.A.; Campagnolo, D.; Gianettoni, J.; Jalili, F.; Kachuck, N.; Lapierre, Y.; et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch. Neurol. 2007, 64, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Von Felten, S.; Traud, S.; Rahman, A.; Quan, J.; King, R.; Garren, H.; Steinman, L.; Cutter, G.; Kappos, L.; et al. Evolution of ms lesions to black holes under DNA vaccine treatment. J. Neurol. 2012, 259, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Selomulya, C.; Ho, J.; Apostolopoulos, V.; Plebanski, M. Delivery of DNA vaccines: An overview on the use of biodegradable polymeric and magnetic nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with plga particles loaded with a mog peptide and il-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghobashy, M.A.; ElMeshad, A.N.; Abdelsalam, R.M.; Nooh, M.M.; Al-Shorbagy, M.; Laible, G. Development and pre-clinical evaluation of recombinant human myelin basic protein nano therapeutic vaccine in experimental autoimmune encephalomyelitis mice animal model. Sci. Rep. 2017, 7, 46468. [Google Scholar] [CrossRef] [PubMed]
- Crowe, P.D.; Qin, Y.; Conlon, P.J.; Antel, J.P. Nbi-5788, an altered mbp83–99 peptide, induces a t-helper 2-like immune response in multiple sclerosis patients. Ann. Neurol. 2000, 48, 758–765. [Google Scholar] [CrossRef]
- Hartung, H.P.; Kieseier, B.C.; Hemmer, B. Purely systemically active anti-inflammatory treatments are adequate to control multiple sclerosis. J. Neurol. 2005, 252, v30–v37. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Comi, G.; Panitch, H.; Oger, J.; Antel, J.; Conlon, P.; Steinman, L.; Comi, G.; Kappos, L.; Oger, J.; et al. Induction of a non-encephalitogenic type 2 t helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase ii trial. Nat. Med. 2000, 6, 1176–1182. [Google Scholar] [PubMed]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase ii clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [PubMed]
- Perera, C.J.; Duffy, S.S.; Lees, J.G.; Kim, C.F.; Cameron, B.; Apostolopoulos, V.; Moalem-Taylor, G. Active immunization with myelin-derived altered peptide ligand reduces mechanical pain hypersensitivity following peripheral nerve injury. J. Neuroinflamm. 2015, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Perera, C.J.; Lees, J.G.; Duffy, S.S.; Makker, P.G.; Fivelman, B.; Apostolopoulos, V.; Moalem-Taylor, G. Effects of active immunisation with myelin basic protein and myelin-derived altered peptide ligand on pain hypersensitivity and neuroinflammation. J. Neuroimmunol. 2015, 286, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.H.; Perera, C.J.; Apostolopoulos, V.; Moalem-Taylor, G. Effects of vaccination with altered peptide ligand on chronic pain in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Front. Neurol. 2013, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Lourbopoulos, A.; Deraos, G.; Matsoukas, M.; Touloumi, O.; Giannakopoulou, A.; Kalbacher, H.; Grigoriadis, N.; Apostolopoulos, V.; Matsoukas, J. Cyclic mog 35–55 ameliorates clinical and neuropathological features of experimental autoimmune encephalomyelitis. Bioorg. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pietersz, G.A.; Loveland, B.E.; Sandrin, M.S.; McKenzie, I.F. Oxidative/reductive conjugation of mannan to antigen selects for t1 or t2 immune responses. Proc. Natl. Acad. Sci. USA 1995, 92, 10128–10132. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pietersz, G.A.; McKenzie, I.F. Cell-mediated immune responses to muc1 fusion protein coupled to mannan. Vaccine 1996, 14, 930–938. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Tsibanis, A.; Tsikkinis, A.; Drakaki, H.; Loveland, B.E.; Piddlesden, S.J.; Plebanski, M.; Pouniotis, D.S.; Alexis, M.N.; et al. Pilot phase iii immunotherapy study in early-stage breast cancer patients using oxidized mannan-muc1 [isrctn71711835]. Breast Cancer Res. 2006, 8, R27. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pietersz, G.A.; Tsibanis, A.; Tsikkinis, A.; Stojanovska, L.; McKenzie, I.F.; Vassilaros, S. Dendritic cell immunotherapy: Clinical outcomes. Clin. Transl. Immunol. 2014, 3, e21. [Google Scholar] [CrossRef] [PubMed]
- Karanikas, V.; Hwang, L.A.; Pearson, J.; Ong, C.S.; Apostolopoulos, V.; Vaughan, H.; Xing, P.X.; Jamieson, G.; Pietersz, G.; Tait, B.; et al. Antibody and t cell responses of patients with adenocarcinoma immunized with mannan-muc1 fusion protein. J. Clin. Investig. 1997, 100, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot phase iii immunotherapy study in stage ii breast cancer patients using oxidized mannan-muc1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-muc1-pulsed dendritic cell immunotherapy: A phase i trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.L.; Quinn, M.A.; Grant, P.T.; Allen, D.G.; Jobling, T.W.; White, S.C.; Zhao, A.; Karanikas, V.; Vaughan, H.; Pietersz, G.; et al. A phase 2, single-arm study of an autologous dendritic cell treatment against mucin 1 in patients with advanced epithelial ovarian cancer. J. Immunother. Cancer 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Day, S.; Tselios, T.; Androutsou, M.E.; Tapeinou, A.; Frilligou, I.; Stojanovska, L.; Matsoukas, J.; Apostolopoulos, V. Mannosylated linear and cyclic single amino acid mutant peptides using a small 10 amino acid linker constitute promising candidates against multiple sclerosis. Front. Immunol. 2015, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Yannakakis, M.P.; Simal, C.; Tzoupis, H.; Rodi, M.; Dargahi, N.; Prakash, M.; Mouzaki, A.; Platts, J.A.; Apostolopoulos, V.; Tselios, T.V. Design and synthesis of non-peptide mimetics mapping the immunodominant myelin basic protein (MBP83–96) epitope to function as t-cell receptor antagonists. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug | Brand | Dose | Number of of Injections, Route | Actions |
|---|---|---|---|---|
| IFN-β1a | Avonex® | 7.5 mg 1st dose | 1/week, i.m | Balances pro- and anti-inflammatory cytokines |
| 15 mg 2nd dose | Decreases Th17 cells | |||
| 22.5 mg 3rd dose | Decreases IL-17 | |||
| 30 mg all subsequent doses | ||||
| Rebif® | 22 mg or 44 mg | 3/week, s.c | ||
| IFN-β1b | Betaseron® | 62.5 mg and increase over 6 weeks to 250 mg | 1/2 days, s.c | |
| Extavia® | 62.5 mg and increase over 6 weeks to 250 mg | 1/2 days, s.c | ||
| pegIFN-β1a | Plegridy® | 63 mg 1st dose | 1/2 weeks, s.c | |
| 95 mg 2nd dose | ||||
| 125 mg all subsequent doses | ||||
| Glatiramer acetate, EKAY | Copaxone® | 20 mg or 40 mg | 1/day, s.c | Blocks pMHC |
| 3/week, s.c | ||||
| Dimethyl fumarate | Tecfidera® | 240 mg | 2–3/day, oral | Anti-inflammatory Anti-oxidative stress |
| Teriflunomide | Aubagio® | 7 or 14 mg | 1/day, oral | Inhibits dihydroorotate dehydrogenase, T, B cells and IFN-γ secreting T cells |
| Fingolimod | Glenya® | 0.5 mg | 1/day, oral | Antagonist of SIP receptor Decrease T, B cells activates SIP signaling in CNS |
| Mitoxantrone | Novatrone® | 12 mg/m2 | 1/3 months up to 2 years | Suppresses T, B cells and macrophages. Reduces Th1 cytokines |
| Dalfampridine | Ampyra® | 10 mg | 2/day, oral | Potassium channel blocker Improves motor symptoms, i.e., walking |
| Humanized Monoclonal Antibody Treatments | ||||
| Natalizumab | Tysabr® | 300 mg | 1/28 days, i.v | Humanized anti-α4-integrin Mab. Affects cell migration, division, growth and survival |
| Ofatumumab | Arzerra® | 3–700 mg | 1/2 weeks, i.v | Humanized anti-CD20 Mab. Cytotoxic to CD20+ cells via CDC and ADCC |
| Ocrelizumab | Ocrevus® | 300–600 mg | 300 mg weeks 1 and 3, then 600 mg 1/6 months, i.v | Humanized anti-CD20 Mab |
| Alemtuzumab | Lemtrada® | 12 mg | 5 days in a row; after 1 year, 3 days | Humanized anti-CD52 Mab. Depletes T, B cells, increases Treg, Th2, decrease Th1 cells |
| Daclizumab | Zinbryta® | 150 mg | 1/month, s.c | Humanized anti-CD25 Mab.Blocks IL-2R, decreases T cells, increases NK cells |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.-E.; De Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci. 2017, 7, 78. https://doi.org/10.3390/brainsci7070078
Dargahi N, Katsara M, Tselios T, Androutsou M-E, De Courten M, Matsoukas J, Apostolopoulos V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sciences. 2017; 7(7):78. https://doi.org/10.3390/brainsci7070078
Chicago/Turabian StyleDargahi, Narges, Maria Katsara, Theodore Tselios, Maria-Eleni Androutsou, Maximilian De Courten, John Matsoukas, and Vasso Apostolopoulos. 2017. "Multiple Sclerosis: Immunopathology and Treatment Update" Brain Sciences 7, no. 7: 78. https://doi.org/10.3390/brainsci7070078
APA StyleDargahi, N., Katsara, M., Tselios, T., Androutsou, M.-E., De Courten, M., Matsoukas, J., & Apostolopoulos, V. (2017). Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sciences, 7(7), 78. https://doi.org/10.3390/brainsci7070078