
Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity


Abstract
:1. Introduction
2. Striatal Anatomy
2.1. Fundamental Structure
2.2. Striatal Compartments
3. Striatal Pathology
3.1. Wild-Type and mHtt Interacting Proteins
3.2. Positron Emission Tomography (PET) Imaging Studies
3.3. Striatum-Predominant Neurodegeneration in HD
3.4. Cell Type-Specific Vulnerability
3.4.1. Glutamate Excitotoxity
3.4.2. Mitochondrial Dysfunction
3.5. Striatal Compartment-Specific Degeneration
3.5.1. Striosome vs. Matrix Neurodegeneration
3.5.2. Dopamine Excitotoxicity-Induced Striosomal Cell Vulnerability
3.5.3. The NPY System Exerts Protective Effects on the Matrix Compartment in HD
3.5.4. Other Proteins May Underlie the Differential Excitotoxicity between Striosome and Matrix Compartments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Albin, R.L.; Tagle, D.A. Genetics and molecular biology of huntington’s disease. Trends Neurosci. 1995, 18, 11–14. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Rub, U.; Vonsattel, J.P.; Heinsen, H.; Korf, H.W. The neuropathology of Huntington’S disease: Classical findings, recent developments and correlation to functional neuroanatomy. Adv. Anat. Embryol. Cell Biol. 2015, 217, 1–146. [Google Scholar] [PubMed]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Lievens, J.C. Pink1-induced mitophagy promotes neuroprotection in huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Hirano, A.; Rojas-Corona, R.R. An immunohistochemical investigation of the human neostriatum in huntington’s disease. Ann. Neurol. 1989, 25, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Augood, S.J.; Faull, R.L.; Love, D.R.; Emson, P.C. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade huntington’s disease: A detailed cellular in situ hybridization study. Neuroscience 1996, 72, 1023–1036. [Google Scholar] [CrossRef]
- Morton, A.J.; Nicholson, L.F.; Faull, R.L. Compartmental loss of nadph diaphorase in the neuropil of the human striatum in huntington’s disease. Neuroscience 1993, 53, 159–168. [Google Scholar] [CrossRef]
- Hedreen, J.C.; Folstein, S.E. Early loss of neostriatal striosome neurons in huntington’s disease. J. Neuropathol. Exp. Neurol. 1995, 54, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Tippett, L.J.; Waldvogel, H.J.; Thomas, S.J.; Hogg, V.M.; van Roon-Mom, W.; Synek, B.J.; Graybiel, A.M.; Faull, R.L. Striosomes and mood dysfunction in huntington’s disease. Brain 2007, 130, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Faull, R.L.; Waldvogel, H.J.; Nicholson, L.F.; Synek, B.J. The distribution of gabaa-benzodiazepine receptors in the basal ganglia in huntington’s disease and in the quinolinic acid-lesioned rat. Prog. Brain Res. 1993, 99, 105–123. [Google Scholar] [PubMed]
- Seto-Ohshima, A.; Emson, P.C.; Lawson, E.; Mountjoy, C.Q.; Carrasco, L.H. Loss of matrix calcium-binding protein-containing neurons in huntington’s disease. Lancet 1988, 1, 1252–1255. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Beal, M.F.; Martin, J.B.; Bird, E.D.; Richardson, E.P., Jr. Morphologic and histochemical characteristics of a spared subset of striatal neurons in huntington’s disease. J. Neuropathol. Exp. Neurol. 1987, 46, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D’Amato, C.J.; Penney, J.B.; Young, A.B. Differential loss of striatal projection neurons in huntington disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5733–5737. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Reiner, A.; Anderson, K.D.; Penney, J.B.; Young, A.B. Striatal and nigral neuron subpopulations in rigid huntington’s disease: Implications for the functional anatomy of chorea and rigidity-akinesia. Ann. Neurol. 1990, 27, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Qin, Y.; Young, A.B.; Penney, J.B.; Chesselet, M.F. Preproenkephalin messenger rna-containing neurons in striatum of patients with symptomatic and presymptomatic huntington’s disease: An in situ hybridization study. Ann. Neurol. 1991, 30, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Reiner, A.; Anderson, K.D.; Dure, L.S., 4th; Handelin, B.; Balfour, R.; Whetsell, W.O., Jr.; Penney, J.B.; Young, A.B. Preferential loss of striato-external pallidal projection neurons in presymptomatic huntington’s disease. Ann. Neurol. 1992, 31, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L. Selective neurodegeneration in huntington’s disease. Ann. Neurol. 1995, 38, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Shelby, E.; Wang, H.; Demarch, Z.; Deng, Y.; Guley, N.H.; Hogg, V.; Roxburgh, R.; Tippett, L.J.; Waldvogel, H.J.; et al. Striatal parvalbuminergic neurons are lost in huntington’s disease: Implications for dystonia. Mov. Disord. 2013, 28, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Nagahiro, S.; Kaji, R. Striosome-Matrix Pathology of Dystonias: A New Hypothesis for Dystonia Genesis, 1st ed.; Nova Science Publisher: Hauppauge, NY, USA, 2010. [Google Scholar]
- Graveland, G.A.; DiFiglia, M. The frequency and distribution of medium-sized neurons with indented nuclei in the primate and rodent neostriatum. Brain Res. 1985, 327, 307–311. [Google Scholar] [CrossRef]
- Cicchetti, F.; Prensa, L.; Wu, Y.; Parent, A. Chemical anatomy of striatal interneurons in normal individuals and in patients with huntington’s disease. Brain Res. Brain Res. Rev. 2000, 34, 80–101. [Google Scholar] [CrossRef]
- Tepper, J.M.; Wilson, C.J.; Koos, T. Feedforward and feedback inhibition in neostriatal gabaergic spiny neurons. Brain Res. Rev. 2008, 58, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Tepper, J.M.; Tecuapetla, F.; Koos, T.; Ibanez-Sandoval, O. Heterogeneity and diversity of striatal gabaergic interneurons. Front. Neuroanat. 2010, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- Ibanez-Sandoval, O.; Tecuapetla, F.; Unal, B.; Shah, F.; Koos, T.; Tepper, J.M. Electrophysiological and morphological characteristics and synaptic connectivity of tyrosine hydroxylase-expressing neurons in adult mouse striatum. J. Neurosci. 2010, 30, 6999–7016. [Google Scholar] [CrossRef] [PubMed]
- Koos, T.; Tepper, J.M. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J. Neurosci. 2002, 22, 529–535. [Google Scholar] [PubMed]
- Centonze, D.; Gubellini, P.; Pisani, A.; Bernardi, G.; Calabresi, P. Dopamine, acetylcholine and nitric oxide systems interact to induce corticostriatal synaptic plasticity. Rev. Neurosci. 2003, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.J.; Hersh, L.B.; Saper, C.B. Cholinergic innervation in the human striatum: A three-compartment model. Neuroscience 1996, 74, 67–87. [Google Scholar] [CrossRef]
- Bolam, J.P. Microcircuits of the Striatum; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Goto, S.; Morigaki, R.; Okita, S.; Nagahiro, S.; Kaji, R. Development of a highly sensitive immunohistochemical method to detect neurochemical molecules in formalin-fixed and paraffin-embedded tissues from autopsied human brains. Front. Neuroanat. 2015, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E.; Crutcher, M.D. Functional architecture of basal ganglia circuits: Neural substrates of parallel processing. Trends Neurosci. 1990, 13, 266–271. [Google Scholar] [CrossRef]
- Gerfen, C.R. The neostriatal mosaic: Multiple levels of compartmental organization. Trends Neurosci. 1992, 15, 133–139. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Malenka, R.C. Striatal plasticity and basal ganglia circuit function. Neuron 2008, 60, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Kuroiwa, M.; Miller, D.B.; O’Callaghan, J.P.; Bateup, H.S.; Shuto, T.; Sotogaku, N.; Fukuda, T.; Heintz, N.; Greengard, P.; et al. Distinct roles of PDE4 and PDE10A in the regulation of camp/pka signaling in the striatum. J. Neurosci. 2008, 28, 10460–10471. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Kuroiwa, M.; Shuto, T. Mechanisms for the modulation of dopamine D(1) receptor signaling in striatal neurons. Front. Neuroanat. 2011, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J. Gabaergic inhibition in the neostriatum. Prog. Brain Res. 2007, 160, 91–110. [Google Scholar] [PubMed]
- Tepper, J.M.; Koos, T.; Wilson, C.J. Gabaergic microcircuits in the neostriatum. Trends Neurosci. 2004, 27, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Ragsdale, C.W., Jr. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc. Natl. Acad. Sci. USA 1978, 75, 5723–5726. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990, 13, 244–254. [Google Scholar] [CrossRef]
- Mikula, S.; Parrish, S.K.; Trimmer, J.S.; Jones, E.G. Complete 3D visualization of primate striosomes by KCHIP1 immunostaining. J. Comp. Neurol. 2009, 514, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Moon Edley, S.; Herkenham, M. Comparative development of striatal opiate receptors and dopamine revealed by autoradiography and histofluorescence. Brain Res. 1984, 305, 27–42. [Google Scholar] [CrossRef]
- Fishell, G.; van der Kooy, D. Pattern formation in the striatum: Developmental changes in the distribution of striatonigral neurons. J. Neurosci. 1987, 7, 1969–1978. [Google Scholar] [PubMed]
- Johnston, J.G.; Gerfen, C.R.; Haber, S.N.; van der Kooy, D. Mechanisms of striatal pattern formation: Conservation of mammalian compartmentalization. Brain Res. Dev. Brain Res. 1990, 57, 93–102. [Google Scholar] [CrossRef]
- van der Kooy, D.; Fishell, G. Neuronal birthdate underlies the development of striatal compartments. Brain Res. 1987, 401, 155–161. [Google Scholar] [CrossRef]
- Krushel, L.A.; Connolly, J.A.; van der Kooy, D. Pattern formation in the mammalian forebrain: Patch neurons from the rat striatum selectively reassociate in vitro. Brain Res. Dev. Brain Res. 1989, 47, 137–142. [Google Scholar] [CrossRef]
- Krushel, L.A.; Fishell, G.; Van der Kooy, D. Pattern formation in the mammalian forebrain: Striatal patch and matrix neurons intermix prior to compartment formation. Eur. J. Neurosci. 1995, 7, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.J.; Graybiel, A.M.; Saper, C.B. Neurochemical architecture of the human striatum. J. Comp. Neurol. 1997, 384, 1–25. [Google Scholar] [CrossRef]
- Gerfen, C.R. The neostriatal mosaic. I. Compartmental organization of projections from the striatum to the substantia nigra in the rat. J. Comp. Neurol. 1985, 236, 454–476. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Castellanos, J.; Graybiel, A.M. Evidence that histochemically distinct zones of the primate substantia nigra pars compacta are related to patterned distributions of nigrostriatal projection neurons and striatonigral fibers. Exp. Brain Res. 1989, 74, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, F.; Sohn, J.; Nakano, T.; Furuta, T.; Nakamura, K.C.; Matsuda, W.; Kaneko, T. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: A single neuron-tracing study using a viral vector. Eur. J. Neurosci. 2011, 33, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, C.W., Jr.; Graybiel, A.M. Compartmental organization of the thalamostriatal connection in the cat. J. Comp. Neurol. 1991, 311, 134–167. [Google Scholar] [CrossRef] [PubMed]
- Christoph, G.R.; Leonzio, R.J.; Wilcox, K.S. Stimulation of the lateral habenula inhibits dopamine-containing neurons in the substantia nigra and ventral tegmental area of the rat. J. Neurosci. 1986, 6, 613–619. [Google Scholar] [PubMed]
- Ji, H.; Shepard, P.D. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J. Neurosci. 2007, 27, 6923–6930. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hikosaka, O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 2007, 447, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-Martin, E.S.; Matsumoto, M.; Nakahara, H.; Hikosaka, O. Multiple timescales of memory in lateral habenula and dopamine neurons. Neuron 2010, 67, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R. The neostriatal mosaic: Multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci. 1992, 15, 285–320. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, J.P.; Herkenham, M. Neostriatal projections from individual cortical fields conform to histochemically distinct striatal compartments in the rat. Brain Res. 1986, 365, 397–403. [Google Scholar] [CrossRef]
- Ragsdale, C.W., Jr.; Graybiel, A.M. Fibers from the basolateral nucleus of the amygdala selectively innervate striosomes in the caudate nucleus of the cat. J. Comp. Neurol. 1988, 269, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A. Neurogenetic patterns in the medial limbic cortex of the rat related to anatomical connections with the thalamus and striatum. Exp. Neurol. 1990, 107, 132–142. [Google Scholar] [CrossRef]
- Flaherty, A.W.; Graybiel, A.M. Corticostriatal transformations in the primate somatosensory system. Projections from physiologically mapped body-part representations. J. Neurophysiol. 1991, 66, 1249–1263. [Google Scholar] [PubMed]
- Flaherty, A.W.; Graybiel, A.M. Two input systems for body representations in the primate striatal matrix: Experimental evidence in the squirrel monkey. J. Neurosci. 1993, 13, 1120–1137. [Google Scholar] [PubMed]
- Eblen, F.; Graybiel, A.M. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci. 1995, 15, 5999–6013. [Google Scholar] [PubMed]
- Levesque, M.; Parent, A. Axonal arborization of corticostriatal and corticothalamic fibers arising from prelimbic cortex in the rat. Cereb. Cortex 1998, 8, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pickel, V.M. Dendritic spines containing mu-opioid receptors in rat striatal patches receive asymmetric synapses from prefrontal corticostriatal afferents. J. Comp. Neurol. 1998, 396, 223–237. [Google Scholar] [CrossRef]
- Penny, G.R.; Wilson, C.J.; Kitai, S.T. Relationship of the axonal and dendritic geometry of spiny projection neurons to the compartmental organization of the neostriatum. J. Comp. Neurol. 1988, 269, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.H.; Arbuthnott, G.W.; Baughman, R.W.; Graybiel, A.M. Dendritic domains of medium spiny neurons in the primate striatum: Relationships to striosomal borders. J. Comp. Neurol. 1993, 337, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Baughman, R.W.; Eckenstein, F. Cholinergic neuropil of the striatum observes striosomal boundaries. Nature 1986, 323, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Aosaki, T.; Flaherty, A.W.; Kimura, M. The basal ganglia and adaptive motor control. Science 1994, 265, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Aosaki, T.; Tsubokawa, H.; Ishida, A.; Watanabe, K.; Graybiel, A.M.; Kimura, M. Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. J. Neurosci. 1994, 14, 3969–3984. [Google Scholar] [PubMed]
- Aosaki, T.; Kimura, M.; Graybiel, A.M. Temporal and spatial characteristics of tonically active neurons of the primate’s striatum. J. Neurophysiol. 1995, 73, 1234–1252. [Google Scholar] [PubMed]
- Miura, M.; Masuda, M.; Aosaki, T. Roles of micro-opioid receptors in gabaergic synaptic transmission in the striosome and matrix compartments of the striatum. Mol. Neurobiol. 2008, 37, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Aosaki, T.; Miura, M.; Suzuki, T.; Nishimura, K.; Masuda, M. Acetylcholine-dopamine balance hypothesis in the striatum: An update. Geriatr. Gerontol. Int. 2010, 10, S148–S157. [Google Scholar] [CrossRef] [PubMed]
- Amemori, K.; Gibb, L.G.; Graybiel, A.M. Shifting responsibly: The importance of striatal modularity to reinforcement learning in uncertain environments. Front. Hum. Neurosci. 2011, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Hori, Y.; Minamimoto, T.; Yamada, H.; Matsumoto, N.; Enomoto, K.; Aosaki, T.; Graybiel, A.M.; Kimura, M. Roles of centromedian parafascicular nuclei of thalamus and cholinergic interneurons in the dorsal striatum in associative learning of environmental events. J. Neural Transm. (Vienna) 2017. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Canales, J.J.; Capper-Loup, C. Levodopa-induced dyskinesias and dopamine-dependent stereotypies: A new hypothesis. Trends Neurosci. 2000, 23, S71–S77. [Google Scholar] [CrossRef]
- Graybiel, A.M. Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci. 2008, 31, 359–387. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Gerfen, C.R. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp. Brain Res. 1998, 123, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Capper-Loup, C.; Canales, J.J.; Kadaba, N.; Graybiel, A.M. Concurrent activation of dopamine D1 and D2 receptors is required to evoke neural and behavioral phenotypes of cocaine sensitization. J. Neurosci. 2002, 22, 6218–6227. [Google Scholar] [PubMed]
- Saka, E.; Goodrich, C.; Harlan, P.; Madras, B.K.; Graybiel, A.M. Repetitive behaviors in monkeys are linked to specific striatal activation patterns. J. Neurosci. 2004, 24, 7557–7565. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Lee, L.V.; Munoz, E.L.; Tooyama, I.; Tamiya, G.; Makino, S.; Ando, S.; Dantes, M.B.; Yamada, K.; Matsumoto, S.; et al. Functional anatomy of the basal ganglia in x-linked recessive dystonia-parkinsonism. Ann. Neurol. 2005, 58, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.S.; Virkud, A.; Deisseroth, K.; Graybiel, A.M. Reversible online control of habitual behavior by optogenetic perturbation of medial prefrontal cortex. Proc. Natl. Acad. Sci. USA 2012, 109, 18932–18937. [Google Scholar] [CrossRef] [PubMed]
- Aosaki, T.; Kawaguchi, Y. Actions of substance p on rat neostriatal neurons in vitro. J. Neurosci. 1996, 16, 5141–5153. [Google Scholar] [PubMed]
- Cragg, S.J. Meaningful silences: How dopamine listens to the ach pause. Trends Neurosci. 2006, 29, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Aosaki, T.; Graybiel, A.M.; Kimura, M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science 1994, 265, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kai, L.; Day, M.; Ronesi, J.; Yin, H.H.; Ding, J.; Tkatch, T.; Lovinger, D.M.; Surmeier, D.J. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 2006, 50, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Flajolet, M.; Greengard, P.; Surmeier, D.J. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 2008, 321, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, R.; Okita, S.; Goto, S. Dopamine-induced changes in galphaolf protein levels in striatonigral and striatopallidal medium spiny neurons underlie the genesis of l-dopa-induced dyskinesia in parkinsonian mice. Front. Cell. Neurosci. 2017, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sumi-Ichinose, C.; Kaji, R.; Ikemoto, K.; Nomura, T.; Nagatsu, I.; Ichinose, H.; Ito, M.; Sako, W.; Nagahiro, S.; et al. Differential involvement of striosome and matrix dopamine systems in a transgenic model of dopa-responsive dystonia. Proc. Natl. Acad. Sci. USA 2008, 105, 12551–12556. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Dunn, D.E.; Merali, F.I.; Woodman, B.; Yim, M.; Borkowska, A.E.; Frosch, M.P.; Bates, G.P.; Housman, D.E.; Lo, D.C.; et al. Caldag-gefi down-regulation in the striatum as a neuroprotective change in huntington’s disease. Hum. Mol. Genet. 2010, 19, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Imarisio, S.; Carmichael, J.; Korolchuk, V.; Chen, C.W.; Saiki, S.; Rose, C.; Krishna, G.; Davies, J.E.; Ttofi, E.; Underwood, B.R.; et al. Huntington’s disease: From pathology and genetics to potential therapies. Biochem. J. 2008, 412, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Chighladze, E.; Arbez, N.; Boronina, T.; Herbrich, S.; Cole, R.N.; Ross, C.A. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 2012, 11, 2006–2021. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, D.; Bauer, J.H.; De-Fraja, C.; Conti, L.; Sipione, S.; Sciorati, C.; Clementi, E.; Hackam, A.; Hayden, M.R.; Li, Y.; et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 2000, 20, 3705–3713. [Google Scholar] [PubMed]
- Rigamonti, D.; Sipione, S.; Goffredo, D.; Zuccato, C.; Fossale, E.; Cattaneo, E. Huntingtin’s neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 2001, 276, 14545–14548. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.W.; Brown, R.; Maxwell, M.; Wyttenbach, A.; Rubinsztein, D.C. Wild type huntingtin reduces the cellular toxicity of mutant huntingtin in mammalian cell models of huntington’s disease. J. Med. Genet. 2001, 38, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse huntington’s disease gene homolog hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.G.; Singaraja, R.; Xanthoudakis, S.; Gutekunst, C.A.; Leavitt, B.R.; Metzler, M.; Hackam, A.S.; Tam, J.; Vaillancourt, J.P.; Houtzager, V.; et al. Recruitment and activation of caspase-8 by the huntingtin-interacting protein hip-1 and a novel partner hippi. Nat. Cell Biol. 2002, 4, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leavitt, B.R.; van Raamsdonk, J.M.; Dragatsis, I.; Goldowitz, D.; MacDonald, M.E.; Hayden, M.R.; Friedlander, R.M. Huntingtin inhibits caspase-3 activation. EMBO J. 2006, 25, 5896–5906. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with rest/nrsf to modulate the transcription of nrse-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing bdnf vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyq proteins in drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.R.; Rong, J.; Li, S.H.; Li, X.J. Interaction of huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Agerman, K.; Dobszay, M.B.; Minichiello, L.; Harkany, T.; Ernfors, P. Brain-derived neurotrophic factor selectively regulates dendritogenesis of parvalbumin-containing interneurons in the main olfactory bulb through the plcgamma pathway. J. Neurobiol. 2006, 66, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Pigino, G.; Brady, S.T. Polyglutamine expansion diseases: Failing to deliver. Trends Mol. Med. 2005, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; You, Y.; Kordower, J.H.; Brady, S.T.; Morfini, G.A. Differential vulnerability of neurons in huntington’s disease: The role of cell type-specific features. J. Neurochem. 2010, 113, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the hd mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Brouillet, E.; Jarraya, B.; Bloch, J.; Jan, C.; Shin, M.; Conde, F.; Li, X.J.; Aebischer, P.; Hantraye, P.; et al. Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol. Ther. 2007, 15, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Humbert, S.; Bryson, E.A.; Cordelieres, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The igf-1/akt pathway is neuroprotective in huntington’s disease and involves huntingtin phosphorylation by akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef]
- Luo, S.; Vacher, C.; Davies, J.E.; Rubinsztein, D.C. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: Implications for mutant huntingtin toxicity. J. Cell. Biol. 2005, 169, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Gafni, J.; Torcassi, C.; Cong, X.; Row, R.H.; LaFevre-Bernt, M.A.; Cusack, M.P.; Ratovitski, T.; Hirschhorn, R.; Ross, C.A.; et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 2006, 281, 23686–23697. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.; Gusella, J.F.; Laramie, J.M.; Myers, R.H.; Lesort, M.; et al. Hd cag repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial structural and functional dynamics in huntington’s disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Li, S.; Wang, C.E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Orr, A.L.; Li, S. Impaired mitochondrial trafficking in huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; De Murtas, M.; Pisani, A.; Stefani, A.; Sancesario, G.; Mercuri, N.B.; Bernardi, G. Vulnerability of medium spiny striatal neurons to glutamate: Role of Na+/K+ atpase. Eur. J. Neurosci. 1995, 7, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Roy, O.W.; Cohen, N.R.; Nicoll, J.A. Pathophysiology of dementias and implications for therapy. Indian J. Pathol. Microbiol. 2005, 48, 289–299. [Google Scholar] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; You, Y.M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Bjorkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Niccolini, F.; Politis, M. Current status of pet imaging in huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; De Micco, R.; Niccolini, F.; Politis, M. Molecular imaging markers to track huntington’s disease pathology. Front. Neurol. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Adamowicz, W.O.; Eldred, W.D.; Jakowski, A.B.; Kleiman, R.J.; Morton, D.G.; Stephenson, D.T.; Strick, C.A.; Williams, R.D.; Menniti, F.S. Cellular and subcellular localization of pde10a, a striatum-enriched phosphodiesterase. Neuroscience 2006, 139, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Nagai, Y.; Miyakawa, T.; Shigemoto, R.; Yokoi, M. Increased social interaction in mice deficient of the striatal medium spiny neuron-specific phosphodiesterase 10A2. J. Neurochem. 2008, 105, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Hill Della Puppa, G.; Reidling, J.; Marcora, E.; Thompson, L.M.; Treanor, J. Comparison of phosphodiesterase 10A, dopamine receptors D1 and D2 and dopamine transporter ligand binding in the striatum of the R6/2 and bachd mouse models of huntington’s disease. J. Huntingt. Dis. 2014, 3, 333–341. [Google Scholar]
- Ooms, M.; Rietjens, R.; Rangarajan, J.R.; Vunckx, K.; Valdeolivas, S.; Maes, F.; Himmelreich, U.; Fernandez-Ruiz, J.; Bormans, G.; Van Laere, K.; et al. Early decrease of type 1 cannabinoid receptor binding and phosphodiesterase 10A activity in vivo in R6/2 huntington mice. Neurobiol. Aging 2014, 35, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.; Okumura, K.; Omori, K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both camp and cgmp (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Haider, S.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Piccini, P.; Kapur, S.; Rabiner, E.A.; et al. Altered pde10a expression detectable early before symptomatic onset in huntington’s disease. Brain 2015, 138, 3016–3029. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Niccolini, F.; Haider, S.; Marques, T.R.; Pagano, G.; Coello, C.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of extra-striatal phosphodiesterase 10A expression in early premanifest huntington’s disease gene carriers. J. Neurol. Sci. 2016, 368, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.S.; Barret, O.; Jennings, D.L.; Friedman, J.H.; Tamagnan, G.D.; Thomae, D.; Alagille, D.; Morley, T.J.; Papin, C.; Papapetropoulos, S.; et al. The phosphodiesterase 10 positron emission tomography tracer, [18F]mni-659, as a novel biomarker for early huntington disease. JAMA Neurol. 2014, 71, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Giampa, C.; Patassini, S.; Borreca, A.; Laurenti, D.; Marullo, F.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of huntington’s disease. Neurobiol. Dis. 2009, 34, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Giampa, C.; Laurenti, D.; Anzilotti, S.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Inhibition of the striatal specific phosphodiesterase pde10A ameliorates striatal and cortical pathology in R6/2 mouse model of huntington’s disease. PLoS ONE 2010, 5, e13417. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; Sammut, S.; Menniti, F.S.; Schmidt, C.J.; West, A.R. Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to cortical stimulation. J. Pharmacol. Exp. Ther. 2009, 328, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Pavese, N.; Tai, Y.F.; Kiferle, L.; Mason, S.L.; Brooks, D.J.; Tabrizi, S.J.; Barker, R.A.; Piccini, P. Microglial activation in regions related to cognitive function predicts disease onset in huntington’s disease: A multimodal imaging study. Hum. Brain Mapp. 2011, 32, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Andrews, T.C.; Weeks, R.A.; Turjanski, N.; Gunn, R.N.; Watkins, L.H.; Sahakian, B.; Hodges, J.R.; Rosser, A.E.; Wood, N.W.; Brooks, D.J. Huntington’s disease progression. Pet and clinical observations. Brain 1999, 122, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Van Oostrom, J.C.; Maguire, R.P.; Verschuuren-Bemelmans, C.C.; Veenma-van der Duin, L.; Pruim, J.; Roos, R.A.; Leenders, K.L. Striatal dopamine D2 receptors, metabolism, and volume in preclinical huntington disease. Neurology 2005, 65, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Leenders, K.L.; Spiegel, R.; Meier, D.; Vontobel, P.; Weigell-Weber, M.; Sanchez-Pernaute, R.; de Yebenez, J.G.; Boesiger, P.; Weindl, A.; et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with huntington’s disease. Brain 1996, 119, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Kassubek, J.; Bernhard Landwehrmeyer, G.; Ecker, D.; Juengling, F.D.; Muche, R.; Schuller, S.; Weindl, A.; Peinemann, A. Global cerebral atrophy in early stages of huntington’s disease: Quantitative mri study. Neuroreport 2004, 15, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Gaura, V.; Ribeiro, M.J.; Lethimonnier, F.; Maroy, R.; Verny, C.; Krystkowiak, P.; Damier, P.; Bachoud-Levi, A.C.; Hantraye, P.; et al. Distribution of grey matter atrophy in huntington’s disease patients: A combined roi-based and voxel-based morphometric study. Neuroimage 2006, 32, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Yasoshima, Y.; Matsushita, N.; Kaneko, T.; Kohno, K.; Pastan, I.; Kobayashi, K. Conditional ablation of striatal neuronal types containing dopamine D2 receptor disturbs coordination of basal ganglia function. J. Neurosci. 2003, 23, 9078–9088. [Google Scholar] [PubMed]
- Li, S.H.; Schilling, G.; Young, W.S., 3rd; Li, X.J.; Margolis, R.L.; Stine, O.C.; Wagster, M.V.; Abbott, M.H.; Franz, M.L.; Ranen, N.G.; et al. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron 1993, 11, 985–993. [Google Scholar] [CrossRef]
- Mestre, T.A.; Sampaio, C. Huntington disease: Linking pathogenesis to the development of experimental therapeutics. Curr. Neurol. Neurosci. Rep. 2017, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Shelbourne, P.F.; Keller-McGandy, C.; Bi, W.L.; Yoon, S.R.; Dubeau, L.; Veitch, N.J.; Vonsattel, J.P.; Wexler, N.S.; Group, U.S.-V.C.R.; Arnheim, N.; et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in huntington disease brain. Hum. Mol. Genet. 2007, 16, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Evans, E.; Chen, C.M.; Craven, L.; Detloff, P.J.; Ennis, M.; Shelbourne, P.F. Dramatic tissue-specific mutation length increases are an early molecular event in huntington disease pathogenesis. Hum. Mol. Genet. 2003, 12, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Furtado, S.; Suchowersky, O.; Rewcastle, B.; Graham, L.; Klimek, M.L.; Garber, A. Relationship between trinucleotide repeats and neuropathological changes in huntington’s disease. Ann. Neurol. 1996, 39, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated bdnf gene transcription in huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; MacDonald, M.E.; McPhaul, M.J.; et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 2003, 40, 41–52. [Google Scholar] [CrossRef]
- Her, L.S.; Goldstein, L.S. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J. Neurosci. 2008, 28, 13662–13672. [Google Scholar] [CrossRef] [PubMed]
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global deprivation of brain-derived neurotrophic factor in the cns reveals an area-specific requirement for dendritic growth. J. Neurosci. 2010, 30, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martin-Ibanez, R.; Munoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. Sumo modification of huntingtin and huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Baiamonte, B.A.; Lee, F.A.; Brewer, S.T.; Spano, D.; LaHoste, G.J. Attenuation of rhes activity significantly delays the appearance of behavioral symptoms in a mouse model of huntington’s disease. PLoS ONE 2013, 8, e53606. [Google Scholar] [CrossRef] [PubMed]
- Mealer, R.G.; Subramaniam, S.; Snyder, S.H. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of huntington’s disease. J. Neurosci. 2013, 33, 4206–4210. [Google Scholar] [CrossRef] [PubMed]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, S. Proteasomal dysfunction in aging and huntington disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sowada, M.J.; Boudreau, R.L.; Aerts, A.M.; Thedens, D.R.; Nopoulos, P.; Davidson, B.L. Rhes suppression enhances disease phenotypes in huntington’s disease mice. J. Huntingt. Dis. 2014, 3, 65–71. [Google Scholar]
- Ferrante, R.J.; Kowall, N.W.; Beal, M.F.; Richardson, E.P., Jr.; Bird, E.D.; Martin, J.B. Selective sparing of a class of striatal neurons in huntington’s disease. Science 1985, 230, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Beal, M.F.; Kowall, N.W.; Richardson, E.P., Jr.; Martin, J.B. Sparing of acetylcholinesterase-containing striatal neurons in huntington’s disease. Brain Res. 1987, 411, 162–166. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef]
- Sharp, A.H.; Loev, S.J.; Schilling, G.; Li, S.H.; Li, X.J.; Bao, J.; Wagster, M.V.; Kotzuk, J.A.; Steiner, J.P.; Lo, A.; et al. Widespread expression of huntington’s disease gene (IT15) protein product. Neuron 1995, 14, 1065–1074. [Google Scholar] [CrossRef]
- Bhide, P.G.; Day, M.; Sapp, E.; Schwarz, C.; Sheth, A.; Kim, J.; Young, A.B.; Penney, J.; Golden, J.; Aronin, N.; et al. Expression of normal and mutant huntingtin in the developing brain. J. Neurosci. 1996, 16, 5523–5535. [Google Scholar] [PubMed]
- Gourfinkel-An, I.; Cancel, G.; Trottier, Y.; Devys, D.; Tora, L.; Lutz, Y.; Imbert, G.; Saudou, F.; Stevanin, G.; Agid, Y.; et al. Differential distribution of the normal and mutated forms of huntingtin in the human brain. Ann. Neurol. 1997, 42, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Landwehrmeyer, G.B.; McNeil, S.M.; Dure, L.S.t.; Ge, P.; Aizawa, H.; Huang, Q.; Ambrose, C.M.; Duyao, M.P.; Bird, E.D.; Bonilla, E.; et al. Huntington’s disease gene: Regional and cellular expression in brain of normal and affected individuals. Ann. Neurol. 1995, 37, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Schilling, G.; Sharp, A.H.; Loev, S.J.; Wagster, M.V.; Li, S.H.; Stine, O.C.; Ross, C.A. Expression of the huntington’s disease (IT15) protein product in hd patients. Hum. Mol. Genet. 1995, 4, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Trottier, Y.; Devys, D.; Imbert, G.; Saudou, F.; An, I.; Lutz, Y.; Weber, C.; Agid, Y.; Hirsch, E.C.; Mandel, J.L. Cellular localization of the huntington’s disease protein and discrimination of the normal and mutated form. Nat. Genet. 1995, 10, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Schwarz, C.; Chase, K.; Bhide, P.G.; Young, A.B.; Penney, J.; Vonsattel, J.P.; Aronin, N.; DiFiglia, M. Huntingtin localization in brains of normal and huntington’s disease patients. Ann. Neurol. 1997, 42, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Senut, M.C.; Suhr, S.T.; Kaspar, B.; Gage, F.H. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J. Neurosci. 2000, 20, 219–229. [Google Scholar] [PubMed]
- De Almeida, L.P.; Ross, C.A.; Zala, D.; Aebischer, P.; Deglon, N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002, 22, 3473–3483. [Google Scholar] [PubMed]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Pisani, A.; Mercuri, N.B.; Bernardi, G. The corticostriatal projection: From synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci. 1996, 19, 19–24. [Google Scholar] [CrossRef]
- Calabresi, P.; Centonze, D.; Pisani, A.; Sancesario, G.; Gubellini, P.; Marfia, G.A.; Bernardi, G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: Implications for ischemia and huntington’s disease. Ann. Neurol. 1998, 43, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Centonze, D.; Pisani, A.; Bernardi, G. Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: Implication for ischemia and huntington’s disease. Exp. Neurol. 1999, 158, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chesselet, M.F.; Gonzales, C.; Lin, C.S.; Polsky, K.; Jin, B.K. Ischemic damage in the striatum of adult gerbils: Relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp. Neurol. 1990, 110, 209–218. [Google Scholar] [CrossRef]
- DiFiglia, M. Excitotoxic injury of the neostriatum: A model for huntington’s disease. Trends Neurosci. 1990, 13, 286–289. [Google Scholar] [CrossRef]
- Goto, S.; Kawarai, T.; Morigaki, R.; Okita, S.; Koizumi, H.; Nagahiro, S.; Munoz, E.L.; Lee, L.V.; Kaji, R. Defects in the striatal neuropeptide y system in x-linked dystonia-parkinsonism. Brain 2013, 136, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Wagster, M.V.; Hedreen, J.C.; Peyser, C.E.; Folstein, S.E.; Ross, C.A. Selective loss of [3H]kainic acid and [3H]AMPA binding in layer vi of frontal cortex in huntington’s disease. Exp. Neurol. 1994, 127, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Young, A.B.; Greenamyre, J.T.; Hollingsworth, Z.; Albin, R.; D’Amato, C.; Shoulson, I.; Penney, J.B. Nmda receptor losses in putamen from patients with huntington’s disease. Science 1988, 241, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Frey, A.S.; Alsdorf, S.A.; Kerner, J.A.; Kosinski, C.M.; Mangiarini, L.; Penney, J.B., Jr.; Davies, S.W.; Bates, G.P.; Young, A.B. Altered neurotransmitter receptor expression in transgenic mouse models of huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Nicniocaill, B.; Haraldsson, B.; Hansson, O.; O’Connor, W.T.; Brundin, P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in r6/1 huntington transgenic mice. Eur. J. Neurosci. 2001, 13, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wyman, T.; Yu, Z.X.; Li, S.H.; Li, X.J. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum. Mol. Genet. 2003, 12, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Klapstein, G.J.; Koppel, A.; Gruen, E.; Cepeda, C.; Vargas, M.E.; Jokel, E.S.; Carpenter, E.M.; Zanjani, H.; Hurst, R.S.; et al. Enhanced sensitivity to n-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of huntington’s disease. J. Neurosci. Res. 1999, 58, 515–532. [Google Scholar] [CrossRef]
- Cepeda, C.; Ariano, M.A.; Calvert, C.R.; Flores-Hernandez, J.; Chandler, S.H.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. Nmda receptor function in mouse models of huntington disease. J. Neurosci. Res. 2001, 66, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.J.; Andre, V.M.; Cepeda, C.; de Lima, M.; Chandler, S.H.; Levine, M.S. Alterations in N-methyl-D-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of huntington’s disease. J. Neurosci. Res. 2005, 82, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.; Le Bourdelles, B.; Heavens, R.P.; Kelly, S.; Smith, D.; Butler, A.; Hammans, R.; Hills, R.; Xuereb, J.H.; Hill, R.G.; et al. The messenger rnas for the N-methyl-D-aspartate receptor subunits show region-specific expression of different subunit composition in the human brain. Neuroscience 1996, 73, 429–447. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Standaert, D.G.; Testa, C.M.; Penney, J.B., Jr.; Young, A.B. Nmda receptor subunit mrna expression by projection neurons and interneurons in rat striatum. J. Neurosci. 1995, 15, 5297–5307. [Google Scholar] [PubMed]
- Kuppenbender, K.D.; Standaert, D.G.; Feuerstein, T.J.; Penney, J.B., Jr.; Young, A.B.; Landwehrmeyer, G.B. Expression of nmda receptor subunit mrnas in neurochemically identified projection and interneurons in the human striatum. J. Comp. Neurol. 2000, 419, 407–421. [Google Scholar] [CrossRef]
- Schoepfer, R.; Monyer, H.; Sommer, B.; Wisden, W.; Sprengel, R.; Kuner, T.; Lomeli, H.; Herb, A.; Kohler, M.; Burnashev, N.; et al. Molecular biology of glutamate receptors. Prog. Neurobiol. 1994, 42, 353–357. [Google Scholar] [CrossRef]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic nmda receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic nmda receptor signaling and expression contributes to phenotype onset in huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: Insights from huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Dau, A.; Gladding, C.M.; Sepers, M.D.; Raymond, L.A. Chronic blockade of extrasynaptic nmda receptors ameliorates synaptic dysfunction and pro-death signaling in huntington disease transgenic mice. Neurobiol. Dis. 2014, 62, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R.; Baimbridge, K.G.; Miller, J.J. The neostriatal mosaic: Compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc. Natl. Acad. Sci. USA 1985, 82, 8780–8784. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, D.; Sapp, E.; Aizawa, H.; Ge, P.; Bird, E.D.; Vonsattel, J.P.; DiFiglia, M. Quinolinic acid-induced increases in calbindin D28K immunoreactivity in rat striatal neurons in vivo and in vitro mimic the pattern seen in huntington’s disease. Neuroscience 1995, 65, 397–407. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, H.B.; Deng, Y.P.; Lei, W.L.; Xie, J.P.; Meade, C.A.; Del Mar, N.; Goldowitz, D.; Reiner, A. Increased calbindin-D28K immunoreactivity in striatal projection neurons of R6/2 huntington’s disease transgenic mice. Neurobiol. Dis. 2005, 20, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.P.; Shelby, E.; Reiner, A.J. Immunohistochemical localization of ampa-type glutamate receptor subunits in the striatum of rhesus monkey. Brain Res. 2010, 1344, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Okita, S.; Morigaki, R.; Koizumi, H.; Kaji, R.; Nagahiro, S.; Goto, S. Cell type-specific localization of optineurin in the striatal neurons of mice: Implications for neuronal vulnerability in huntington’s disease. Neuroscience 2012, 202, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Savanenin, A.; Reddy, P.H.; Liu, Y.F. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J. Biol. Chem. 2001, 276, 24713–24718. [Google Scholar] [CrossRef] [PubMed]
- Anborgh, P.H.; Godin, C.; Pampillo, M.; Dhami, G.K.; Dale, L.B.; Cregan, S.P.; Truant, R.; Ferguson, S.S. Inhibition of metabotropic glutamate receptor signaling by the huntingtin-binding protein optineurin. J. Biol. Chem. 2005, 280, 34840–34848. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Kawaguchi, Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J. Neurosci. 1996, 16, 2397–2410. [Google Scholar] [PubMed]
- Lee, J.M.; Ivanova, E.V.; Seong, I.S.; Cashorali, T.; Kohane, I.; Gusella, J.F.; MacDonald, M.E. Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet. 2007, 3, e135. [Google Scholar] [CrossRef] [PubMed]
- Medina, L.; Figueredo-Cardenas, G.; Reiner, A. Differential abundance of superoxide dismutase in interneurons versus projection neurons and in matrix versus striosome neurons in monkey striatum. Brain Res. 1996, 708, 59–70. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Antioxidants in huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Kowall, N.W. Tyrosine hydroxylase-like immunoreactivity is distributed in the matrix compartment of normal human and huntington’s disease striatum. Brain Res. 1987, 416, 141–146. [Google Scholar] [CrossRef]
- Lee, J.M.; Ramos, E.M.; Lee, J.H.; Gillis, T.; Mysore, J.S.; Hayden, M.R.; Warby, S.C.; Morrison, P.; Nance, M.; Ross, C.A.; et al. Cag repeat expansion in huntington disease determines age at onset in a fully dominant fashion. Neurology 2012, 78, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Keum, J.W.; Shin, A.; Gillis, T.; Mysore, J.S.; Abu Elneel, K.; Lucente, D.; Hadzi, T.; Holmans, P.; Jones, L.; Orth, M.; et al. The htt cag-expansion mutation determines age at death but not disease duration in huntington disease. Am. J. Hum. Genet. 2016, 98, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; Baimbridge, K.G. Relative loss of the striatal striosome compartment, defined by calbindin-D28K immunostaining, following developmental hypoxic-ischemic injury. Neuroscience 1993, 56, 305–315. [Google Scholar] [CrossRef]
- Fuchs, T.; Saunders-Pullman, R.; Masuho, I.; Luciano, M.S.; Raymond, D.; Factor, S.; Lang, A.E.; Liang, T.W.; Trosch, R.M.; White, S.; et al. Mutations in gnal cause primary torsion dystonia. Nat. Genet. 2013, 45, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kaji, R.; Matsumoto, S.; Nagahiro, S.; Goto, S. Compartmental loss of striatal medium spiny neurons in multiple system atrophy of parkinsonian type. Mov. Disord. 2007, 22, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Lawhorn, C.; Smith, D.M.; Brown, L.L. Striosome-matrix pathology and motor deficits in the YAC128 mouse model of huntington’s disease. Neurobiol. Dis. 2008, 32, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Graybiel, A.M. Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 2011, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.C.; Garas, S.N.; Drouin-Ouellet, J.; Mason, S.L.; Stott, S.R.; Barker, R.A. Dopamine and huntington’s disease. Expert Rev. Neurother. 2015, 15, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Andre, V.M.; Cepeda, C.; Levine, M.S. Dopamine and glutamate in huntington’s disease: A balancing act. CNS Neurosci. Ther. 2010, 16, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Spokes, E.G. Neurochemical alterations in huntington’s chorea: A study of post-mortem brain tissue. Brain 1980, 103, 179–210. [Google Scholar] [CrossRef] [PubMed]
- Garrett, M.C.; Soares-da-Silva, P. Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in huntington’s disease: Evidence for an overactive dopaminergic brain transmission. J. Neurochem. 1992, 58, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yohrling, G.J., 4th; Jiang, G.C.; DeJohn, M.M.; Miller, D.W.; Young, A.B.; Vrana, K.E.; Cha, J.H. Analysis of cellular, transgenic and human models of huntington’s disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res. Mol. Brain Res. 2003, 119, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Jakel, R.J.; Maragos, W.F. Neuronal cell death in huntington’s disease: A potential role for dopamine. Trends Neurosci. 2000, 23, 239–245. [Google Scholar] [CrossRef]
- Hickey, M.A.; Reynolds, G.P.; Morton, A.J. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of huntington’s disease. J. Neurochem. 2002, 81, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Garrett, N.J. Striatal dopamine and homovanillic acid in huntington’s disease. J. Neural Transm. 1986, 65, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Borrelli, E. Dopamine in neurotoxicity and neuroprotection: What do D2 receptors have to do with it? Trends Neurosci. 2006, 29, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sako, W.; Morigaki, R.; Nagahiro, S.; Kaji, R.; Goto, S. Olfactory type g-protein alpha subunit in striosome-matrix dopamine systems in adult mice. Neuroscience 2010, 170, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Thapliyal, A.; Bannister, R.A.; Hanks, C.; Adams, B.A. The monomeric g proteins AGS1 and rhes selectively influence galphai-dependent signaling to modulate N-type (CAV2.2) calcium channels. Am. J. Physiol. Cell Physiol. 2008, 295, C1417–C1426. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; He, Y. Rhes and AGS1/Dexras1 affect signaling by dopamine D1 receptors through adenylyl cyclase. J. Neurosci. Res. 2011, 89, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, P.; De Abajo, R.; Garcia-Ranea, J.A.; Valencia, A.; Santisteban, P.; Crespo, P.; Bernal, J. The small gtp-binding protein, rhes, regulates signal transduction from g protein-coupled receptors. Oncogene 2004, 23, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Santini, E.; Migliarini, S.; Borgkvist, A.; Centonze, D.; Nasti, V.; Carta, M.; De Chiara, V.; Prosperetti, C.; Spano, D.; et al. The gtp-binding protein rhes modulates dopamine signalling in striatal medium spiny neurons. Mol. Cell Neurosci. 2008, 37, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Napolitano, F.; Pelosi, B.; Schepisi, C.; Migliarini, S.; Di Maio, A.; Pendolino, V.; Mancini, M.; Sciamanna, G.; Vitucci, D.; et al. Rhes influences striatal cAMP/PKA-dependent signaling and synaptic plasticity in a gender-sensitive fashion. Sci. Rep. 2015, 5, 10933. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A receptors in the brain: Current research and their role in neurodegeneration. Molecules 2017, 22, E676. [Google Scholar] [CrossRef] [PubMed]
- Dawbarn, D.; De Quidt, M.E.; Emson, P.C. Survival of basal ganglia neuropeptide y-somatostatin neurones in huntington’s disease. Brain Res. 1985, 340, 251–260. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Bjorklund, A. Comparison of the behavioural and histological characteristics of the 6-ohda and alpha-synuclein rat models of parkinson’s disease. Exp. Neurol. 2012, 235, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta 1998, 1366, 211–223. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Hayden, M.R. Deranged neuronal calcium signaling and huntington disease. Biochem. Biophys. Res. Commun. 2004, 322, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.S.; Chen, X.; Liu, J.; Bezprozvanny, I. Dopaminergic signaling and striatal neurodegeneration in huntington’s disease. J. Neurosci. 2007, 27, 7899–7910. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, R.; Goto, S. Postsynaptic density protein 95 in the striosome and matrix compartments of the human neostriatum. Front. Neuroanat. 2015, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Greengard, P. The neurobiology of dopamine signaling. Biosci. Rep. 2001, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Morigaki, R.; Sako, W.; Okita, S.; Kasahara, J.; Yokoyama, H.; Nagahiro, S.; Kaji, R.; Goto, S. Cyclin-dependent kinase 5 with phosphorylation of tyrosine 15 residue is enriched in striatal matrix compartment in adult mice. Neuroscience 2011, 189, 25–31. [Google Scholar] [CrossRef] [PubMed]
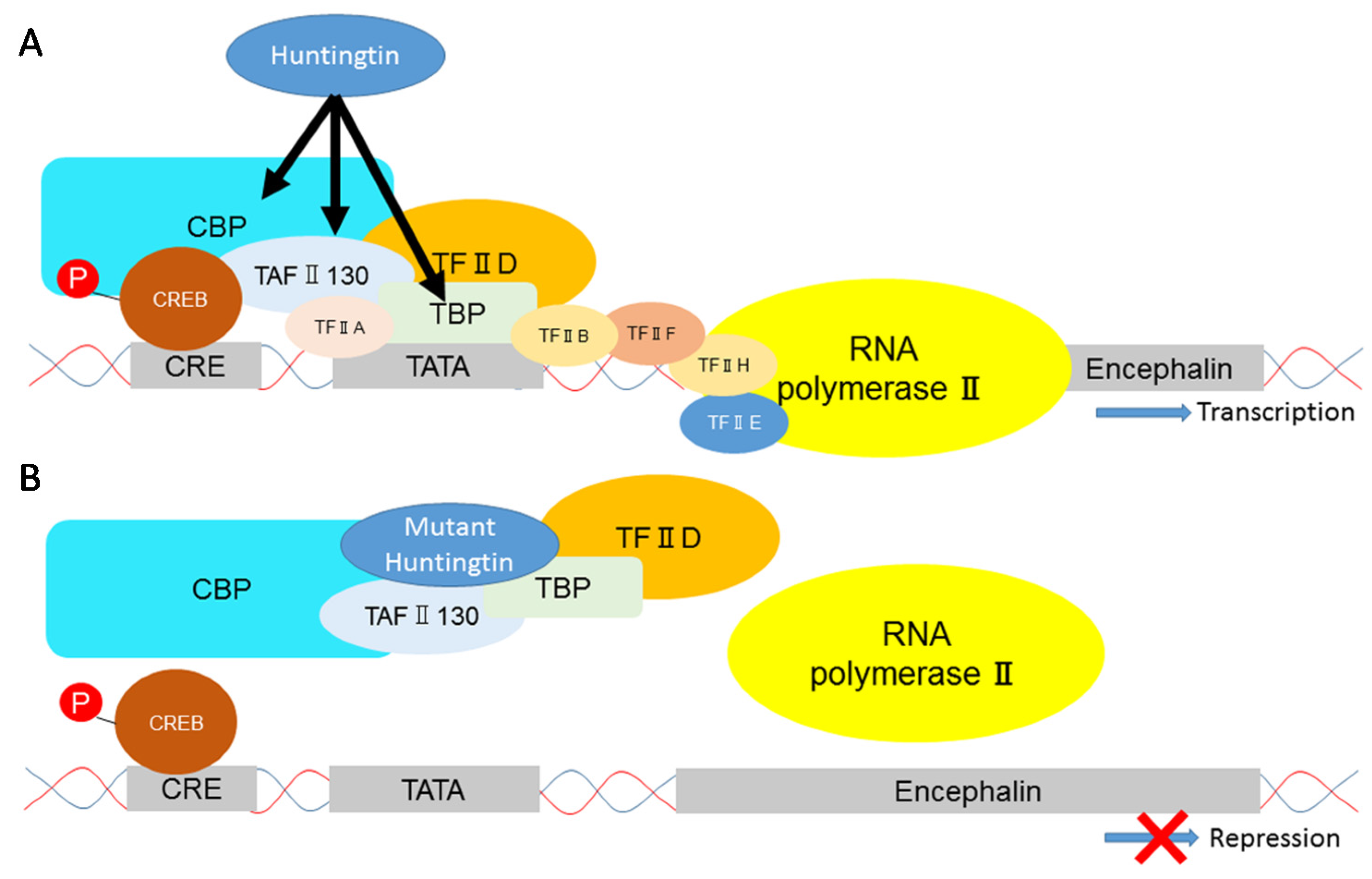

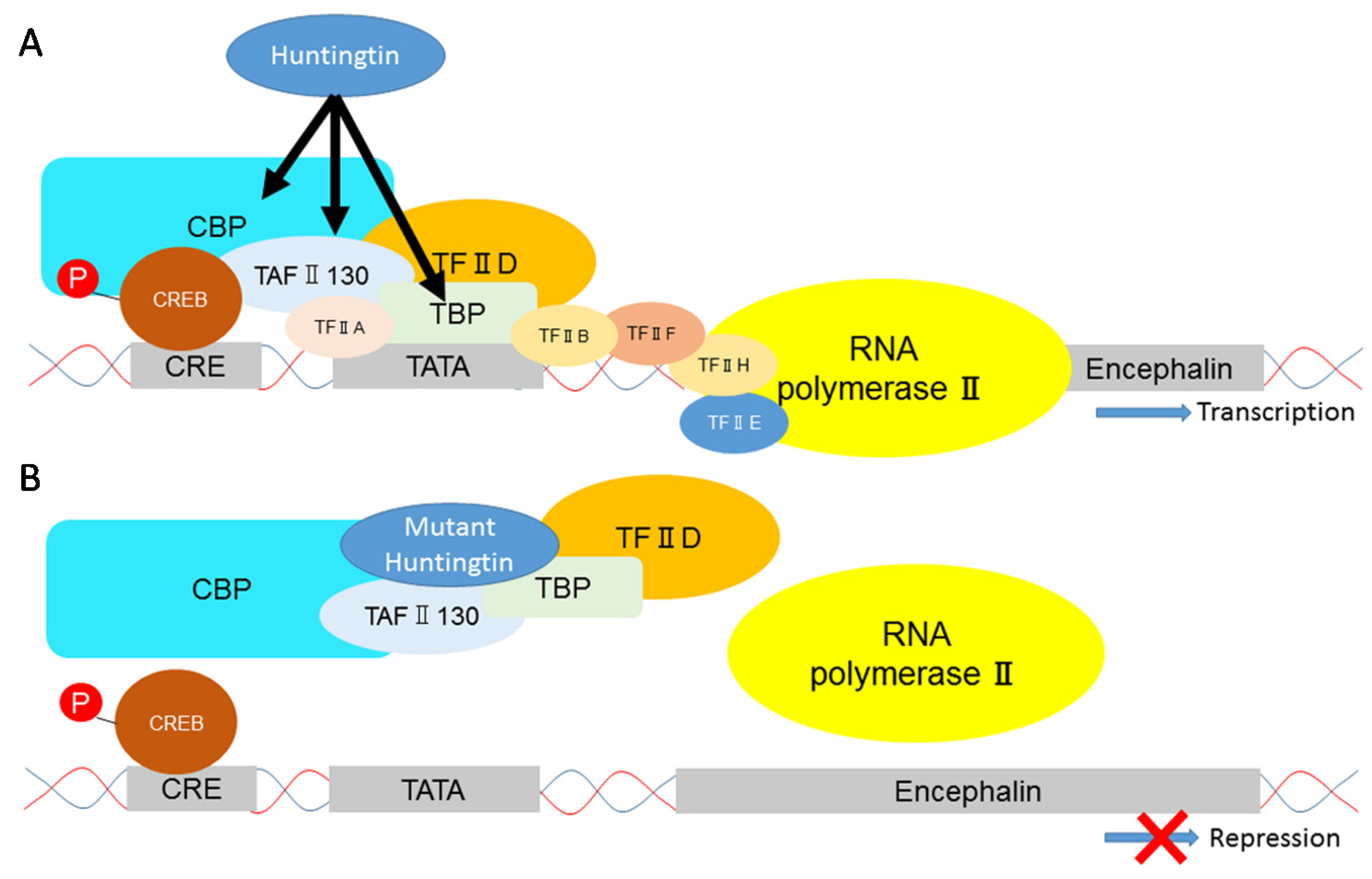{kind=link}
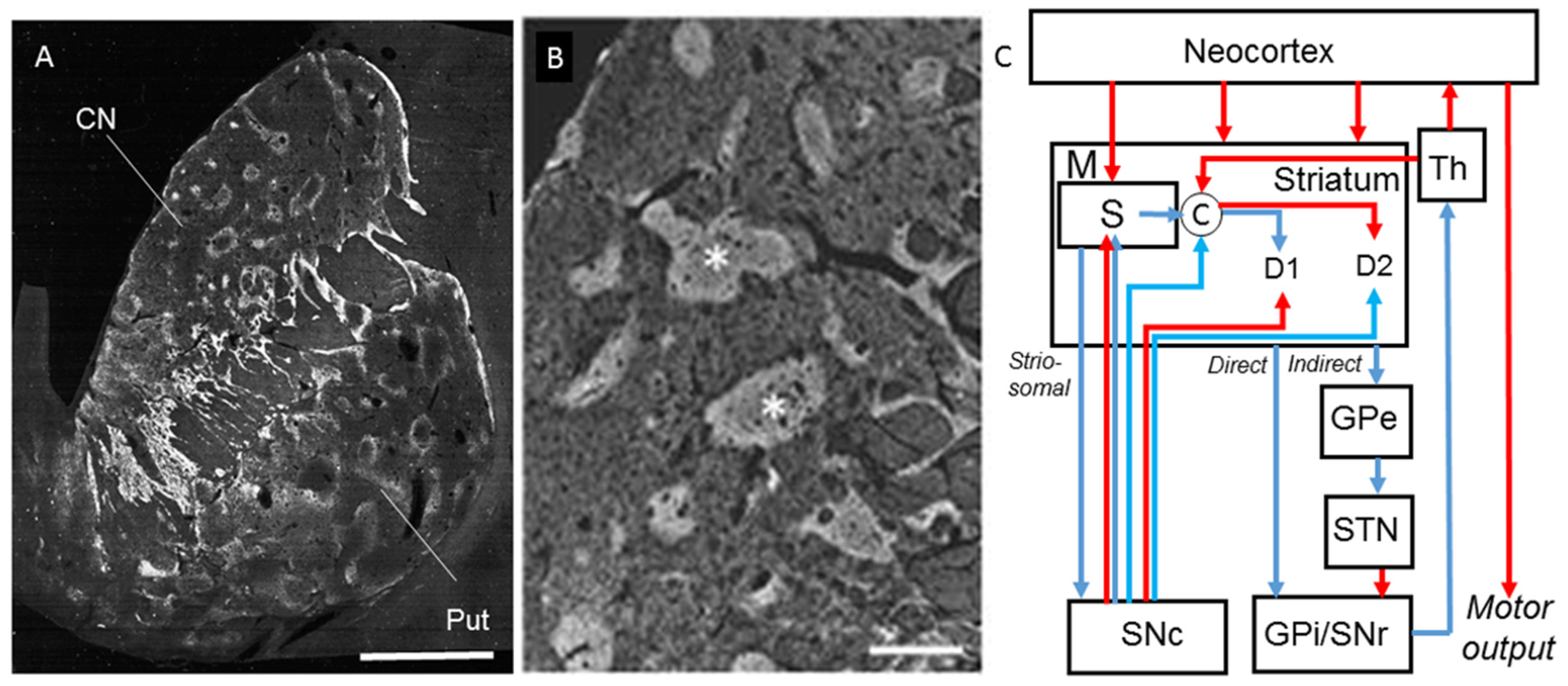
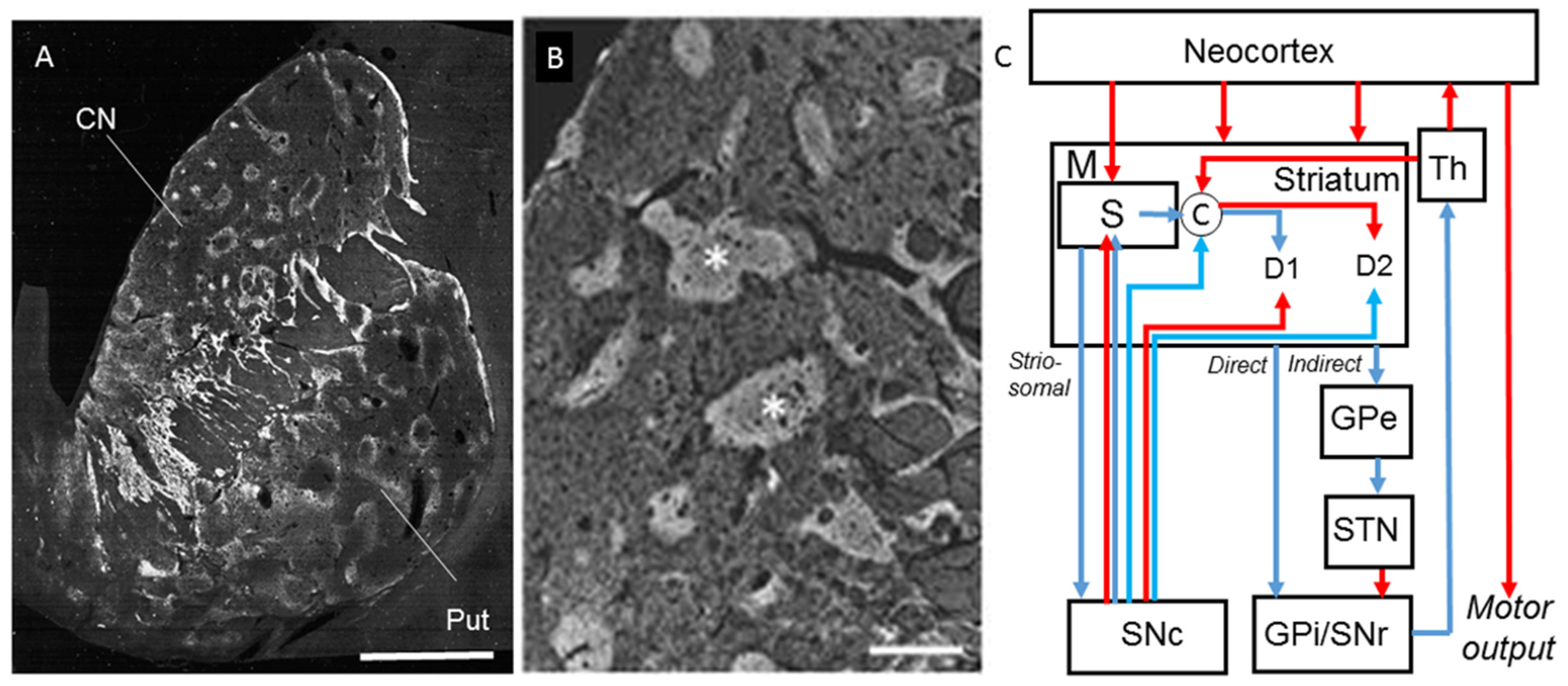{kind=link}
| Affected Structures or Cell-Types | Neuroprotective or Neurotoxic Effects | Factors | Hypothesized Mechanism |
|---|---|---|---|
| Striatum | Neurotoxic or neuroprotective | Predominant expression of Ras homolog enriched in striatum (Rhes) in the striatum | Neurotoxic: Rhes binds mHtt and increases cytotoxicity or decreases the autophagic capacity of the striatal neurons. |
| Neuroprotective: Rhes activates autophagy of mHtt and induces gene expression promoting mHtt degradation. | |||
| MSNs | Neurotoxic | Brain-derived neurotrophic factor (BDNF) deletion in HD | BDNF is required for survival and dendritic growth of MSNs preferentially in indirect pathway. |
| N-methyl D-aspartate receptor subtype 2B (NR2B) predominant expression in MSNs | High expression of NR2B in MSNs may promote NMDA excitotoxicity. | ||
| Sensitivity to glutamate | MSNs are more sensitive to kainite, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA), N-methyl D-aspartate receptor (NMDA), and group 1 metabotropic glutamate receptor (mGluR) agonists than cholinergic interneurons. | ||
| Increased NR2B containing extrasynaptic NMDA receptors | Extrasynaptic NMDA receptors increase the toxic mHtt and cause dysregulation in p38 mitogen-activated protein kinase-cAMP response element binding protein (MAPK-CREB) signaling. | ||
| Requirement of higher energy in MSNs | Susceptibility for mitochondrial dysfunction induced by mutant-Htt. | ||
| Expression level of superoxide dismutases (SODs) | MSNs contain low level of SODs, which indicates the vulnerability against oxidative stress. | ||
| Neuroprotective | Increased expression of parvalbumin (PVA), calretinin, and calcium binding proteins | Calcium-buffering effect over excessive calcium-induced excitotoxicity. | |
| PDE10A deletion | Activation of extracellular signal-regulated kinase (ERK), CREB and predominant activation of D2R indirect pathway. | ||
| PVAs | Neurotoxic | BDNF deletion | Blockade of tropomyosin receptor kinase B-phospholipase Cγ (TrkB-PLCγ) pathway by BDNF deletion. |
| AMPA receptors expression | Enrichment in Ca2+ permeable AMPA receptors induced calcium-induced excitotoxicity. | ||
| Interneurons | Neuroprotective | Optineurin expression | Optineurin is predominantly expressed in striatal interneurons and negatively regulates glutamate receptors via interaction with Htt. |
| Striosomes | Neurotoxic | Phosphodiesterase 10A (PDE-10A) decrease in cortical regions projecting to striosomes | PDE-10A decrease induces neurodegeneration in cortical neurons projecting to striosomes. |
| Dopamine D1R | Enrichment of D1R in striosomes induces dopamine excitotoxicity. | ||
| SOD2 | Lower expression level of SOD2 in striosomes induces vulnerability against oxidative stress. | ||
| Olfactory type G-protein α subunit (Gαolf) | Enrichment of Gαolf in striosomes induces D1R and A2AR mediated neurotoxicity (decreased level of Rhes increases A2AR/cAMP/protein kinase A (PKA) activity). | ||
| Matrix | Neuroprotective | Cyclin-dependent kinase 5 (CDK5) | Matrix enrich protein CDK5-pY15 induces phosphorylation of Htt which results in decrease of toxic effects against mutant-Htt. |
| Decrease of calcium diacylglycerol guanine nucleotide exchange factor 1 (CalDAG-GEF1) | Matrix and MSNs predominant expression pattern of CalDAG-GEF1 protects them from mutant-Htt induced toxicity. | ||
| Dopamine D2R | Enrichment of D2R in matrix is protective against dopamine excitotoxicity. | ||
| Neuropeptide Y (NPY) | Enrichment of NPY in matrix is protective against glutamate excitotoxicity and microglial activation. | ||
| 28 kDa calbindin (Calbindin-D28K) | Enrichment of Calbindin-D28K in matrix is protective against excessive Ca2+ influx by calcium-buffering. | ||
| Post synaptic density 95 kDa (PSD-95) | Matrix enrich protein PSD-95 is protective against glutamatergic or dopaminergic excitotoxicity. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morigaki, R.; Goto, S. Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity. Brain Sci. 2017, 7, 63. https://doi.org/10.3390/brainsci7060063
Morigaki R, Goto S. Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity. Brain Sciences. 2017; 7(6):63. https://doi.org/10.3390/brainsci7060063
Chicago/Turabian StyleMorigaki, Ryoma, and Satoshi Goto. 2017. "Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity" Brain Sciences 7, no. 6: 63. https://doi.org/10.3390/brainsci7060063
APA StyleMorigaki, R., & Goto, S. (2017). Striatal Vulnerability in Huntington’s Disease: Neuroprotection Versus Neurotoxicity. Brain Sciences, 7(6), 63. https://doi.org/10.3390/brainsci7060063