
Microarray Analysis Reveals Higher Gestational Folic Acid Alters Expression of Genes in the Cerebellum of Mice Offspring—A Pilot Study

Abstract
:1. Introduction
2. Results
2.1. Higher Maternal FA during Gestation Alters Expression of Several Genes in the Cerebellum of Offspring
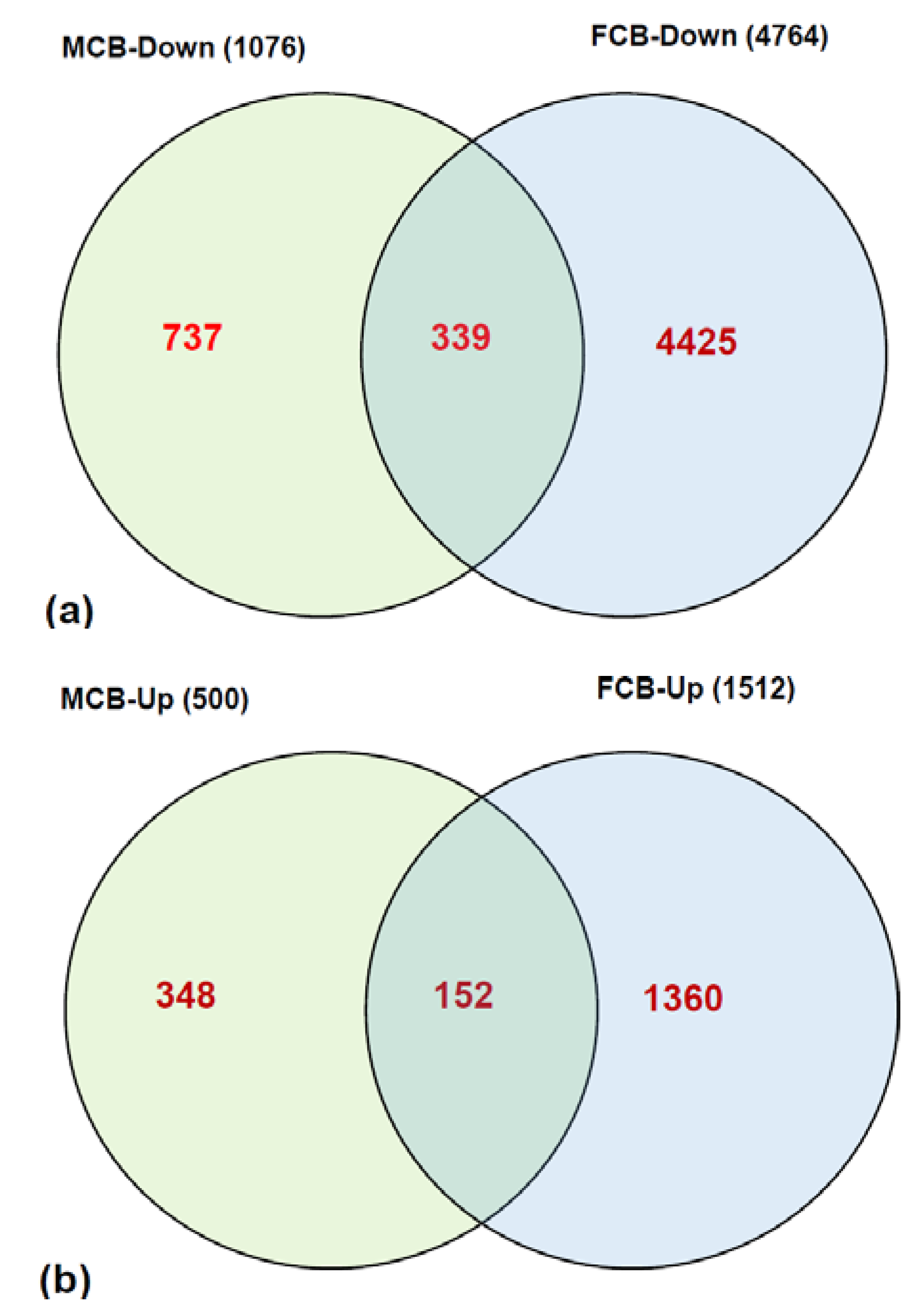
2.2. Higher Maternal FA Alters Expression of Several Transcription Factors and Imprinted Genes in the Cerebellum of Offspring
2.3. Higher Maternal FA Alters the Expression Profile of Several Neural Genes in the Cerebellum of Offspring

{kind=link}
{kind=link}
| Accession | Symbol | Fold Change | Direction of Change | Pathway |
|---|---|---|---|---|
| ref|NM_146087 | Csnk1a1 | −3.90 | ↓ | Circadian |
| ref|NM_007771 | Cry1 | −2.70 | ↓ | Circadian |
| ref|NM_172523 | Slc18a2 | −3.81 | ↓ | Dopa-Serotonin |
| ref|NM_201607 | Pde4c | −3.11 | ↓ | Dopa-Serotonin |
| ref|NM_010484 | Slc6a4 | −3.06 | ↓ | Dopa-Serotonin |
| ref|NM_008740 | Nsf | −3.40 | ↓ | GABA-glutamate |
| ref|NM_001130444 | Hras1 | −4.63 | ↓ | Gap-Junctions |
| ref|NM_009446 | Tuba3a | −3.17 | ↓ | Gap-Junctions |
| ref|NM_023279 | Tubb3 | −2.76 | ↓ | Gap-Junctions |
| ref|NM_023716 | Tubb2b | −2.71 | ↓ | Gap-Junctions |
| ref|NM_007659 | Cdk1 | −2.53 | ↓ | Gap-Junctions |
| ref|NM_008235 | Hes1 | −3.17 | ↓ | Neurogenesis |
| ref|NM_001039104 | Trpm1 | −3.36 | ↓ | Neuronal-Ion channel |
| ens|ENSMUST00000105918 | Kcnq1 | −3.29 | ↓ | Neuronal-Ion channel |
| ref|NM_144939 | Frs3 | −6.24 | ↓ | Neurotrophin-receptor |
| ref|NM_053075 | Rheb | −3.13 | ↓ | Synaptic Plasticity |
| ref|NM_138305 | Adcy3 | 2.71 | ↑ | Dopa-Serotonin |
| ref|NM_019840 | Pde4b | 2.57 | ↑ | Dopa-Serotonin |
| ref|NM_009007 | Rac1 | 2.73 | ↑ | Neurogenesis |
| ref|NM_011913 | Best1 | 2.62 | ↑ | Neuronal-Ion channel |
| ref|NM_009314 | Tacr2 | 2.73 | ↑ | Neurotransmitter |
| ref|NM_009750 | Ngfrap1 | 2.92 | ↑ | Neurotrophin-receptor |
| Accession | Symbol | Fold Change | Direction of Change | Pathway |
|---|---|---|---|---|
| ref|NM_146087 | Csnk1a1 | −7.80 | ↓ | Circadian |
| ref|NM_011158 | Prkar2b | −6.84 | ↓ | Circadian |
| ref|NM_013672 | Sp1 | −5.80 | ↓ | Circadian |
| ref|NM_008540 | Smad4 | −5.02 | ↓ | Circadian |
| ref|NM_172563 | Hlf | −4.27 | ↓ | Circadian |
| ref|NM_009602 | Chrnb2 | −4.10 | ↓ | Circadian |
| ref|NM_001174053 | Camk2b | −3.37 | ↓ | Circadian |
| ref|NM_015822 | Fbxl3 | −3.27 | ↓ | Circadian |
| ref|NM_009516 | Wee1 | −3.05 | ↓ | Circadian |
| ref|NM_010098 | Opn3 | −3.04 | ↓ | Circadian |
| ref|NM_008904 | Ppargc1a | −2.90 | ↓ | Circadian |
| ref|NM_011144 | Ppara | −2.56 | ↓ | Circadian |
| ref|NM_013642 | Dusp1 | −4.63 | ↓ | Dopa-Serotonin |
| ref|NM_011866 | Pde10a | −4.55 | ↓ | Dopa-Serotonin |
| ref|NM_010234 | Fos | −4.47 | ↓ | Dopa-Serotonin |
| ref|NM_001131020 | Gfap | −4.29 | ↓ | Dopa-Serotonin |
| ref|NM_001012765 | Adcy5 | −3.83 | ↓ | Dopa-Serotonin |
| ref|NM_172778 | Maob | −3.67 | ↓ | Dopa-Serotonin |
| ref|NM_001111015 | Syn2 | −3.11 | ↓ | Dopa-Serotonin |
| gb|AK032648 | Pde4d | −2.63 | ↓ | Dopa-Serotonin |
| ref|NM_019827 | Gsk3b | −2.55 | ↓ | Dopa-Serotonin |
| ref|NM_009732 | Avp | −13.07 | ↓ | GABA-glutamate |
| ref|NM_007578 | Cacna1a | −8.80 | ↓ | GABA-glutamate |
| ref|NM_008069 | Gabrb1 | −7.02 | ↓ | GABA-glutamate |
| ref|NM_146072 | Grik1 | −5.57 | ↓ | GABA-glutamate |
| ref|NM_001037724 | Adcy7 | −5.50 | ↓ | GABA-glutamate |
| ref|NM_008174 | Grm8 | −5.34 | ↓ | GABA-glutamate |
| ref|NM_008171 | Grin2b | −5.07 | ↓ | GABA-glutamate |
| ref|NM_001039195 | Gria2 | −5.07 | ↓ | GABA-glutamate |
| ref|NM_001146311 | Cln3 | −5.01 | ↓ | GABA-glutamate |
| ref|NM_001013385 | Grm4 | −4.57 | ↓ | GABA-glutamate |
| ref|NM_176942 | Gabra5 | −4.20 | ↓ | GABA-glutamate |
| ref|NM_010251 | Gabra4 | −4.12 | ↓ | GABA-glutamate |
| ref|NM_011393 | Slc1a2 | −3.99 | ↓ | GABA-glutamate |
| ref|NM_001113383 | Gls | −3.95 | ↓ | GABA-glutamate |
| ref|NM_182959 | Slc17a8 | −3.91 | ↓ | GABA-glutamate |
| ref|NM_016886 | Gria3 | −3.89 | ↓ | GABA-glutamate |
| ref|NM_008075 | Gabrr1 | −3.83 | ↓ | GABA-glutamate |
| ref|NM_080853 | Slc17a6 | −3.49 | ↓ | GABA-glutamate |
| ref|NM_175328 | Slc6a15 | −3.15 | ↓ | GABA-glutamate |
| ref|NM_001143834 | Grm5 | −3.10 | ↓ | GABA-glutamate |
| ref|NM_008740 | Nsf | −3.09 | ↓ | GABA-glutamate |
| ref|NM_019691 | Gria4 | −3.03 | ↓ | GABA-glutamate |
| ref|NM_001042451 | Snca | −2.80 | ↓ | GABA-glutamate |
| ref|NM_147176 | Homer1 | −2.69 | ↓ | GABA-glutamate |
| ref|NM_021284 | Kras | −6.85 | ↓ | Gap-Junctions |
| ref|NM_009446 | Tuba3a | −6.70 | ↓ | Gap-Junctions |
| ref|NM_008927 | Map2k1 | −4.92 | ↓ | Gap-Junctions |
| ref|NM_010937 | Nras | −4.37 | ↓ | Gap-Junctions |
| ref|NM_011101 | Prkca | −3.98 | ↓ | Gap-Junctions |
| ref|NM_007659 | Cdk1 | −3.84 | ↓ | Gap-Junctions |
| ref|NM_001080971 | Tubb1 | −3.67 | ↓ | Gap-Junctions |
| ref|NM_011100 | Prkacb | −3.64 | ↓ | Gap-Junctions |
| ref|NM_009447 | Tuba4a | −3.62 | ↓ | Gap-Junctions |
| ref|NM_010288 | Gja1 | −3.52 | ↓ | Gap-Junctions |
| ref|NM_175452 | Gjc2 | −2.91 | ↓ | Gap-Junctions |
| ref|NM_010930 | Nov | −2.91 | ↓ | Gap-Junctions |
| ref|NM_007912 | Egfr | −2.71 | ↓ | Gap-Junctions |
| ref|NM_009231 | Sos1 | −2.70 | ↓ | Gap-Junctions |
| ref|NM_028751 | Tjap1 | −2.53 | ↓ | Gap-Junctions |
| ref|NM_001025250 | Vegfa | −6.08 | ↓ | Neurogenesis |
| ref|NM_008744 | Ntn1 | −5.16 | ↓ | Neurogenesis |
| ref|NM_007889 | Dvl3 | −4.83 | ↓ | Neurogenesis |
| ref|NM_010894 | Neurod1 | −4.13 | ↓ | Neurogenesis |
| ref|NM_008781 | Pax3 | −4.07 | ↓ | Neurogenesis |
| ref|NM_177821 | Ep300 | −3.87 | ↓ | Neurogenesis |
| ref|NM_011443 | Sox2 | −3.54 | ↓ | Neurogenesis |
| ref|NM_009599 | Ache | −3.50 | ↓ | Neurogenesis |
| ref|NM_033620 | Pard3 | −3.34 | ↓ | Neurogenesis |
| ref|NM_010928 | Notch2 | −3.25 | ↓ | Neurogenesis |
| ref|NM_008737 | Nrp1 | −3.07 | ↓ | Neurogenesis |
| ref|NM_007553 | Bmp2 | −2.86 | ↓ | Neurogenesis |
| ref|NM_022312 | Tnr | −2.85 | ↓ | Neurogenesis |
| ref|NM_010883 | Ndp | −2.62 | ↓ | Neurogenesis |
| ref|NM_001039934 | Mtap2 | −2.60 | ↓ | Neurogenesis |
| ref|NM_007865 | Dll1 | −2.59 | ↓ | Neurogenesis |
| ref|NM_008973 | Ptn | −2.54 | ↓ | Neurogenesis |
| ref|NM_008421 | Kcnc1 | −10.41 | ↓ | Neuronal-Ion channel |
| ref|NM_010597 | Kcnab1 | −5.07 | ↓ | Neuronal-Ion channel |
| ref|NM_009900 | Clcn2 | −3.81 | ↓ | Neuronal-Ion channel |
| ref|NM_001025581 | Kcnc2 | −3.45 | ↓ | Neuronal-Ion channel |
| ref|NM_001083616 | Cacna1d | −3.26 | ↓ | Neuronal-Ion channel |
| ref|NM_008226 | Hcn2 | −3.23 | ↓ | Neuronal-Ion channel |
| ref|NM_011930 | Clcn7 | −3.04 | ↓ | Neuronal-Ion channel |
| ref|NM_010408 | Hcn1 | −2.85 | ↓ | Neuronal-Ion channel |
| ref|NM_145983 | Kcna5 | −2.67 | ↓ | Neuronal-Ion channel |
| ref|NM_001099298 | Scn2a1 | −2.60 | ↓ | Neuronal-Ion channel |
| ref|NM_001044308 | Cacna1i | −2.59 | ↓ | Neuronal-Ion channel |
| ref|NM_018732 | Scn3a | −2.51 | ↓ | Neuronal-Ion channel |
| ref|NM_010610 | Kcnma1 | −2.50 | ↓ | Neuronal-Ion channel |
| ref|NM_153087 | Hrh4 | −4.01 | ↓ | Neurotransmitter |
| ref|NM_001081147 | Oxtr | −3.68 | ↓ | Neurotransmitter |
| ref|NM_008285 | Hrh1 | −2.90 | ↓ | Neurotransmitter |
| ref|NM_021382 | Tacr3 | −2.71 | ↓ | Neurotransmitter |
| ref|NM_009313 | Tacr1 | −2.70 | ↓ | Neurotransmitter |
| ref|NM_008747 | Ntsr2 | −2.61 | ↓ | Neurotransmitter |
| ref|NM_009219 | Sstr4 | −2.61 | ↓ | Neurotransmitter |
| ref|NM_008177 | Grpr | −2.53 | ↓ | Neurotransmitter |
| ref|NM_001025074 | Ntrk2 | −7.28 | ↓ | Neurotrophin-receptor |
| ref|NM_009365 | Tgfb1i1 | −6.21 | ↓ | Neurotrophin-receptor |
| ref|NM_144939 | Frs3 | −5.29 | ↓ | Neurotrophin-receptor |
| ref|NM_011640 | Trp53 | −4.40 | ↓ | Neurotrophin-receptor |
| ref|NM_010560 | Il6st | −4.00 | ↓ | Neurotrophin-receptor |
| ref|NM_019791 | Maged1 | −3.69 | ↓ | Neurotrophin-receptor |
| ens|ENSMUST00000052164 | Ppyr1 | −3.47 | ↓ | Neurotrophin-receptor |
| ref|NM_009911 | Cxcr4 | −3.39 | ↓ | Neurotrophin-receptor |
| ref|NM_022024 | Gmfg | −3.10 | ↓ | Neurotrophin-receptor |
| ref|NM_010849 | Myc | −3.06 | ↓ | Neurotrophin-receptor |
| ref|NM_139149 | Fus | −2.84 | ↓ | Neurotrophin-receptor |
| ref|NM_177410 | Bcl2 | −2.64 | ↓ | Neurotrophin-receptor |
| ref|NM_001025074 | Ntrk2 | −7.28 | ↓ | Synaptic Plasticity |
| ref|NM_028736 | Grip1 | −6.77 | ↓ | Synaptic Plasticity |
| ref|NM_020493 | Srf | −3.25 | ↓ | Synaptic Plasticity |
| ens|ENSMUST00000111939 | Nos1 | −2.97 | ↓ | Synaptic Plasticity |
| ref|NM_013498 | Crem | −2.88 | ↓ | Synaptic Plasticity |
| ref|NM_009952 | Creb1 | −2.83 | ↓ | Synaptic Plasticity |
| ref|NM_024469 | Bhlhe41 | 5.46 | ↑ | Circadian |
| ref|NM_145712 | Mtnr1b | 4.18 | ↑ | Circadian |
| ref|NM_008679 | Ncoa3 | 3.70 | ↑ | Circadian |
| ref|NM_017376 | Tef | 3.48 | ↑ | Circadian |
| ref|NM_011281 | Rorc | 3.19 | ↑ | Circadian |
| ref|NM_138305 | Adcy3 | 4.41 | ↑ | Dopa-Serotonin |
| ref|NM_008169 | Grin1 | 5.56 | ↑ | GABA-glutamate |
| ref|NM_009200 | Slc1a6 | 2.80 | ↑ | GABA-glutamate |
| ref|NM_008121 | Gja5 | 4.27 | ↑ | Gap-Junctions |
| ref|NM_007616 | Cav1 | 3.57 | ↑ | Gap-Junctions |
| ref|NM_016975 | Gja3 | 2.98 | ↑ | Gap-Junctions |
| ref|NM_011840 | Map2k5 | 2.72 | ↑ | Gap-Junctions |
| ref|NM_008361 | Il1b | 2.67 | ↑ | GABA-glutamate |
| ens|ENSMUST00000114274 | Robo1 | 4.31 | ↑ | Neurogenesis |
| ref|NM_008782 | Pax5 | 3.39 | ↑ | Neurogenesis |
| ref|NM_011486 | Stat3 | 2.92 | ↑ | Neurogenesis |
| ref|NM_001077403 | Nrp2 | 2.52 | ↑ | Neurogenesis |
| ref|NM_183000 | Accn3 | 4.44 | ↑ | Neuronal-Ion channel |
| ref|NM_007699 | Chrm4 | 3.82 | ↑ | Neurotransmitter |
| ref|NM_013462 | Adrb3 | 2.61 | ↑ | Neurotransmitter |
| ref|NM_008006 | Fgf2 | 2.65 | ↑ | Neurotrophin-receptor |
| ref|NM_008416 | Junb | 2.86 | ↑ | Synaptic Plasticity |
| ref|NM_007664 | Cdh2 | 2.82 | ↑ | Synaptic Plasticity |
3. Discussion
4. Material and Methods
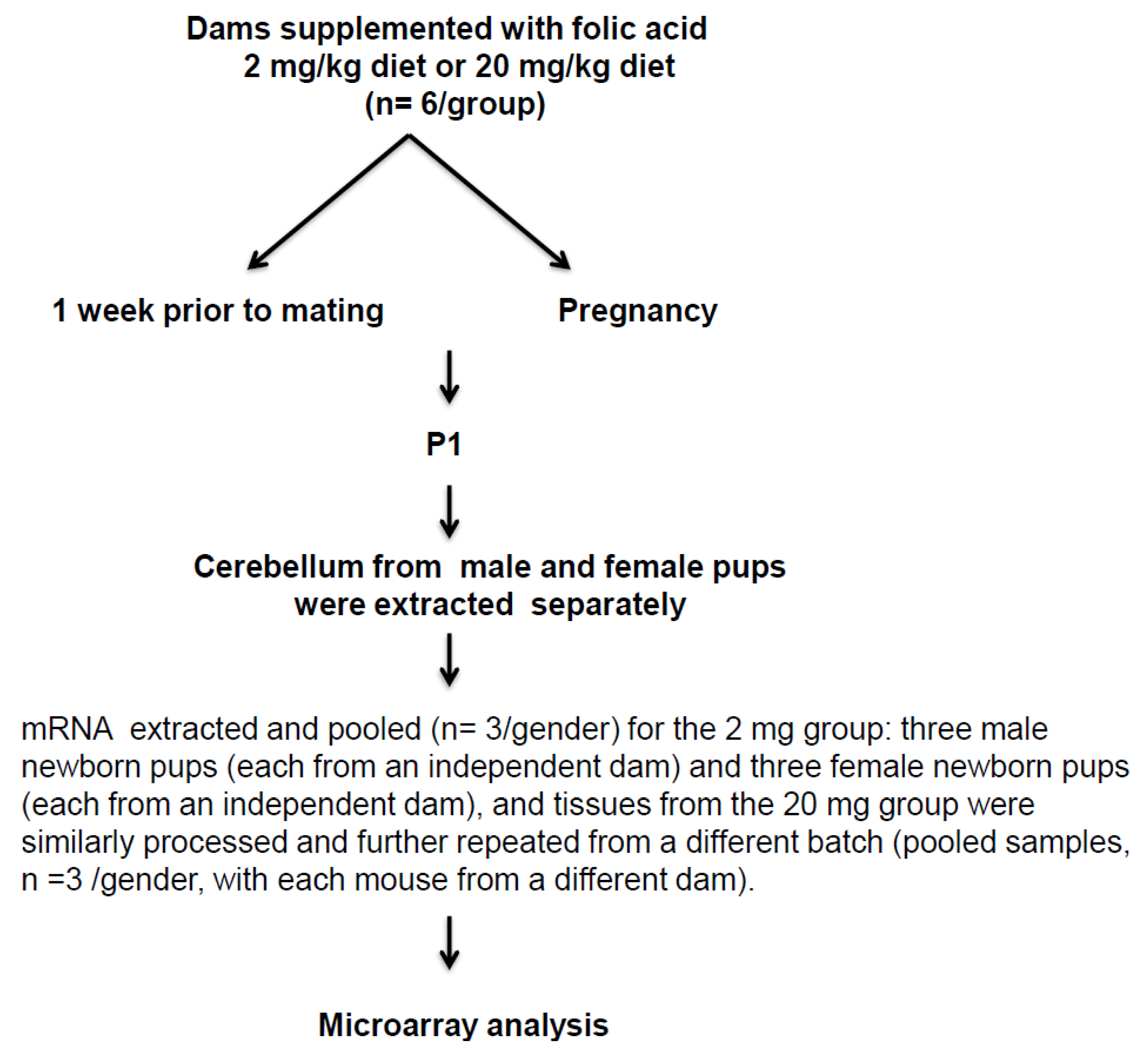
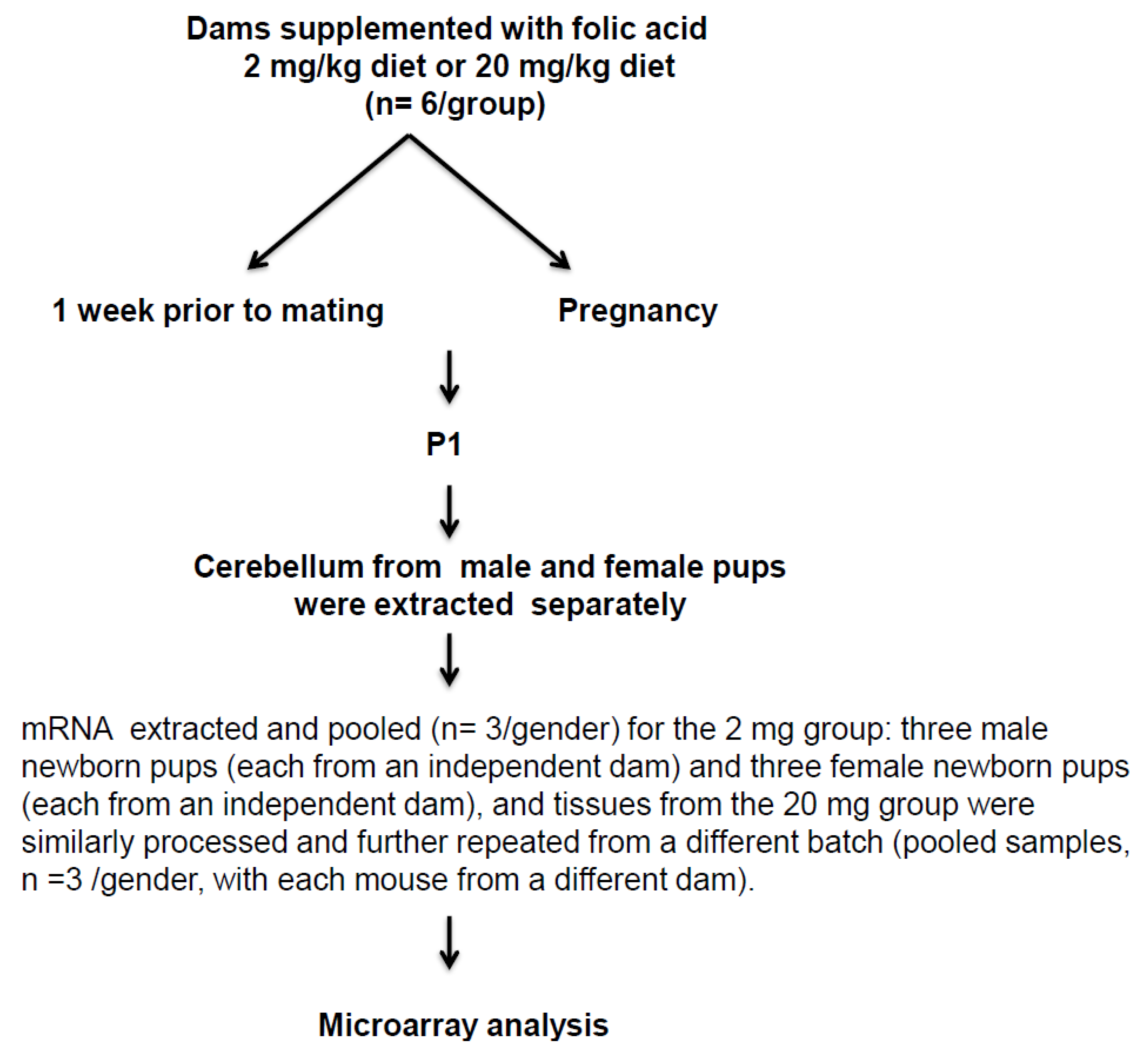
4.1. Mice Strain and Feeding
4.2. RNA Preparation
4.3. Microarrays
4.4. Data Analysis
5. Conclusions
Acknowledgements
Authors Contributions
Supplementary Information
Conflicts of Interest
References
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Vanhees, K.; Vonhogen, I.G.; van Schooten, F.J.; Godschalk, R.W. You are what you eat, and so are your children: The impact of micronutrients on the epigenetic programming of offspring. Cell Mol. Life Sci. 2014, 71, 271–285. [Google Scholar] [CrossRef]
- Callender, S. A critical review of pernicious anaemia of pregnancy. Q. J. Med. 1944, 13, 75–105. [Google Scholar]
- Hibbard, E.D.; Smithells, R.W. Folic acid metabolism and human embryopathy. Lancet 1965, 285. [Google Scholar] [CrossRef]
- Barua, S.; Kuizon, S.; Junaid, M.A. Folic acid supplementation in pregnancy and implications in health and disease. J. Biomed. Sci. 2014, 21. [Google Scholar] [CrossRef]
- Honein, M.A.; Paulozzi, L.J.; Mathews, T.J.; Erickson, J.D.; Wong, L.Y. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA 2001, 285, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- De Wals, P.; Tairou, F.; van Allen, M.I.; Uh, S.H.; Lowry, R.B.; Sibbald, B.; Evans, J.A.; van den Hof, M.C.; Zimmer, P.; Crowley, M.; et al. Reduction in neural-tube defects after folic acid fortification in Canada. In N. Engl. J. Med.; 2007; Volume 357, pp. 135–142. [Google Scholar]
- U.S. Preventive Services Task Force. Folic acid for the prevention of neural tube defects: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2009, 150, 626–631. [Google Scholar]
- Choumenkovitch, S.F.; Selhub, J.; Wilson, P.W.; Rader, J.I.; Rosenberg, I.H.; Jacques, P.F. Folic acid intake from fortification in United States exceeds predictions. J. Nutr. 2002, 132, 2792–2798. [Google Scholar]
- Barua, S.; Kuizon, S.; Chadman, K.K.; Flory, M.J.; Brown, W.T.; Junaid, M.A. Single-base resolution of mouse offspring brain methylome reveals epigenome modifications caused by gestational folic acid. Epigenetics Chromatin 2014, 7. [Google Scholar] [CrossRef]
- Main, P.A.; Angley, M.T.; Thomas, P.; O’Doherty, C.E.; Fenech, M. Folate and methionine metabolism in autism: A systematic review. Am. J. Clin. Nutr. 2010, 91, 1598–1620. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Kloth, A.D.; Badura, A. The Cerebellum, Sensitive Periods, and Autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Reeber, S.L.; Otis, T.S.; Sillitoe, R.V. New roles for the cerebellum in health and disease. Front. Syst. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Wisniewski, T.; Brown, W. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef]
- Horsthemke, B. In brief: Genomic imprinting and imprinting diseases. J. Pathol. 2014, 232, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Tranebjaerg, L.; Bathen, J.; Tyson, J.; Bitner-Glindzicz, M. Jervell and Lange-Nielsen syndrome: A Norwegian perspective. Am. J. Med. Genet. 1999, 89, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Celano, G.; Dagradi, F.; Schwartz, P.J. Congenital long QT syndrome. Orphanet J. Rare Dis. 2008, 3. [Google Scholar] [CrossRef]
- Senner, C.E.; Brockdorff, N. Xist gene regulation at the onset of X inactivation. Curr. Opin. Genet. Dev. 2009, 19, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Plagge, A. Non-Coding RNAs at the Gnas and Snrpn-Ube3a Imprinted Gene Loci and Their Involvement in Hereditary Disorders. Front. Genet. 2012, 3. [Google Scholar] [CrossRef]
- Riccio, A.; Sparago, A.; Verde, G.; de Crescenzo, A.; Citro, V.; Cubellis, M.V.; Ferrero, G.B.; Silengo, M.C.; Russo, S.; Larizza, L.; Cerrato, F. Inherited and Sporadic Epimutations at the IGF2-H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumor. Endocr. Dev. 2009, 14, 1–9. [Google Scholar]
- Soejima, H.; Higashimoto, K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J. Hum. Genet. 2013, 58, 402–409. [Google Scholar]
- Antypa, N.; Serretti, A.; Rujescu, D. Serotonergic genes and suicide: A systematic review. Eur. Neuropsychopharmacol. 2013, 23, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.Y. Transcriptional dysregulation of neocortical circuit assembly in ASD. Int. Rev. Neurobiol. 2013, 113, 167–205. [Google Scholar] [PubMed]
- Cukier, H.N.; Rabionet, R.; Konidari, I.; Rayner-Evans, M.Y.; Baltos, M.L.; Wright, H.H.; Abramson, R.K.; Martin, E.R.; Cuccaro, M.L.; Pericak-Vance, M.A.; Gilbert, J.R. Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics 2010, 11, 291–303. [Google Scholar] [CrossRef]
- Vijayendran, M.; Beach, S.R.; Plume, J.M.; Brody, G.H.; Philibert, R.A. Effects of genotype and child abuse on DNA methylation and gene expression at the serotonin transporter. Front. Psychiatry 2012, 3. [Google Scholar] [CrossRef]
- Huang, C.H.; Santangelo, S.L. Autism and serotonin transporter gene polymorphisms: A systematic review and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 903–913. [Google Scholar] [CrossRef]
- Oberlander, T.F. Fetal serotonin signaling: Setting pathways for early childhood development and behavior. J. Adolesc. Health 2012, 51 (Suppl. S2), 9–16. [Google Scholar] [CrossRef]
- Dannlowski, U.; Kugel, H.; Redlich, R.; Halik, A.; Schneider, I.; Opel, N.; Grotegerd, D.; Schwarte, K.; Schettler, C.; Ambree, O.; et al. Serotonin transporter gene methylation is associated with hippocampal gray matter volume. Hum. Brain Map. 2014, 35, 5356–5367. [Google Scholar] [CrossRef]
- Tabolacci, E.; Chiurazzi, P. Epigenetics, fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. A 2013, 161A, 2797–2808. [Google Scholar] [CrossRef]
- Brown, W.T. Clinical aspects of the fragile X syndrome. Results Probl. Cell Differ. 2012, 54, 273–279. [Google Scholar] [PubMed]
- Tsiouris, J.A.; Brown, W.T. Neuropsychiatric symptoms of fragile X syndrome: Pathophysiology and pharmacotherapy. CNS Drugs 2004, 18, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.A.; Kuizon, S.; Cardona, J.; Azher, T.; Murakami, N.; Pullarkat, R.K.; Brown, W.T. Folic acid supplementation dysregulates gene expression in lymphoblastoid cells—Implications in nutrition. Biochem. Biophys. Res. Commun. 2011, 412, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.C.; Bailey, D.B., Jr.; Berry-Kravis, E.; Greenberg, J.; Losh, M.; Mailick, M.; Mila, M.; Olichney, J.M.; Rodriguez-Revenga, L.; Sherman, S.; et al. Associated features in females with an FMR1 premutation. J. Neurodev. Disord. 2014, 6. [Google Scholar] [CrossRef]
- Yadav, R.; Gupta, S.C.; Hillman, B.G.; Bhatt, J.M.; Stairs, D.J.; Dravid, S.M. Deletion of glutamate delta-1 receptor in mouse leads to aberrant emotional and social behaviors. PLoS One 2012, 7, e32969. [Google Scholar] [CrossRef]
- Wu, S.; Yue, W.; Jia, M.; Ruan, Y.; Lu, T.; Gong, X.; Shuang, M.; Liu, J.; Yang, X.; Zhang, D. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 492–495. [Google Scholar] [CrossRef]
- Barua, S.; Chadman, K.K.; Kuizon, S.; Buenaventura, D.; Stapley, N.W.; Ruocco, F.; Begum, U.; Guariglia, S.R.; Brown, W.T.; Junaid, M.A. Increasing Maternal or Post-Weaning Folic Acid Alters Gene Expression and Moderately Changes Behavior in the Offspring. PLoS One 2014, 9, e101674. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.R.; McPartlin, J.; Weir, D.G.; Daly, S.; Pentieva, K.; Daly, L.; Scott, J.M. Evidence of unmetabolised folic acid in cord blood of newborn and serum of 4-day-old infants. Br. J. Nutr. 2005, 94, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Kim, Y.I.; Refsum, H. Is folic acid good for everyone? Am. J. Clin. Nutr. 2008, 87, 517–533. [Google Scholar] [PubMed]
- Rogers, E.J. Has enhanced folate status during pregnancy altered natural selection and possibly Autism prevalence? A closer look at a possible link. Med. Hypotheses 2008, 71, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Leeming, R.J.; Lucock, M. Autism: Is there a folate connection? J. Inherit. Metab. Dis. 2009, 32, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.M.; Panser, L.A.; Katusic, S.K. Is excess folic acid supplementation a risk factor for autism? Med. Hypotheses 2011, 77, 15–17. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.J.; Vrana, P.B.; Rosenfeld, C.S. Maternal methyl supplemented diets and effects on offspring health. Front. Genet. 2014, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Neggers, Y.H. Increasing prevalence, changes in diagnostic criteria, and nutritional risk factors for autism spectrum disorders. ISRN Nutr. 2014, 2014. [Google Scholar] [CrossRef]
- Cahill, L. Why sex matters for neuroscience. Nat. Rev. Neurosci. 2006, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.P.; Mazure, C.M.; Staley, J.K. Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol. Psychiatry 2007, 62, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Jazin, E.; Cahill, L. Sex differences in molecular neuroscience: From fruit flies to humans. Nat. Rev. Neurosci. 2010, 11, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Periconceptional influences on offspring sex ratio and placental responses. Reprod. Fertil. Dev. 2011, 24, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Trabzuni, D.; Ramasamy, A.; Imran, S.; Walker, R.; Smith, C.; Weale, M.E.; Hardy, J.; Ryten, M. Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Galanopoulou, A.S. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 2008, 80, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [PubMed]
- Deghan, M.S.; Ishiguro, L.; Sohn, K.J.; Medline, A.; Renlund, R.; Croxford, R.; Kim, Y.I. Folic acid supplementation promotes mammary tumor progression in a rat model. PLoS One 2014, 9, e84635. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Lam, A.; Li, M.C.; Ngan, M.; Menenzes, S.; Zhao, Y. Analysis of gene expression data using BRB-Array Tools. Cancer Inform. 2007, 3, 11–17. [Google Scholar] [PubMed]
- Shorter, K.R.; Anderson, V.; Cakora, P.; Owen, A.; Lo, K.; Crossland, J.; South, A.C.; Felder, M.R.; Vrana, P.B. Pleiotropic effects of a methyl donor diet in a novel animal model. PLoS One 2014, 9, e104942. [Google Scholar] [CrossRef] [PubMed]
- Dwarkanath, P.; Barzilay, J.R.; Thomas, T.; Thomas, A.; Bhat, S.; Kurpad, A.V. High folate and low vitamin B-12 intakes during pregnancy are associated with small-for-gestational age infants in South Indian women: A prospective observational cohort study. Am. J. Clin. Nutr. 2013, 98, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D. Folic acid fortification: The good, the bad, and the puzzle of vitamin B-12. Am. J. Clin. Nutr. 2007, 85, 3–5. [Google Scholar] [PubMed]
- Black, M.M. Effects of vitamin B12 and folate deficiency on brain development in children. Food Nutr. Bull. 2008, 29 (Suppl. S2), 126–131. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barua, S.; Kuizon, S.; Chadman, K.K.; Brown, W.T.; Junaid, M.A. Microarray Analysis Reveals Higher Gestational Folic Acid Alters Expression of Genes in the Cerebellum of Mice Offspring—A Pilot Study. Brain Sci. 2015, 5, 14-31. https://doi.org/10.3390/brainsci5010014
Barua S, Kuizon S, Chadman KK, Brown WT, Junaid MA. Microarray Analysis Reveals Higher Gestational Folic Acid Alters Expression of Genes in the Cerebellum of Mice Offspring—A Pilot Study. Brain Sciences. 2015; 5(1):14-31. https://doi.org/10.3390/brainsci5010014
Chicago/Turabian StyleBarua, Subit, Salomon Kuizon, Kathryn K. Chadman, W. Ted Brown, and Mohammed A. Junaid. 2015. "Microarray Analysis Reveals Higher Gestational Folic Acid Alters Expression of Genes in the Cerebellum of Mice Offspring—A Pilot Study" Brain Sciences 5, no. 1: 14-31. https://doi.org/10.3390/brainsci5010014
APA StyleBarua, S., Kuizon, S., Chadman, K. K., Brown, W. T., & Junaid, M. A. (2015). Microarray Analysis Reveals Higher Gestational Folic Acid Alters Expression of Genes in the Cerebellum of Mice Offspring—A Pilot Study. Brain Sciences, 5(1), 14-31. https://doi.org/10.3390/brainsci5010014