Reprogramming Cells for Brain Repair

Abstract
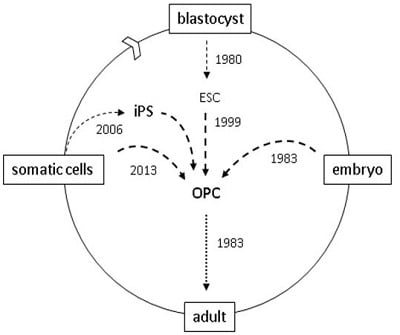
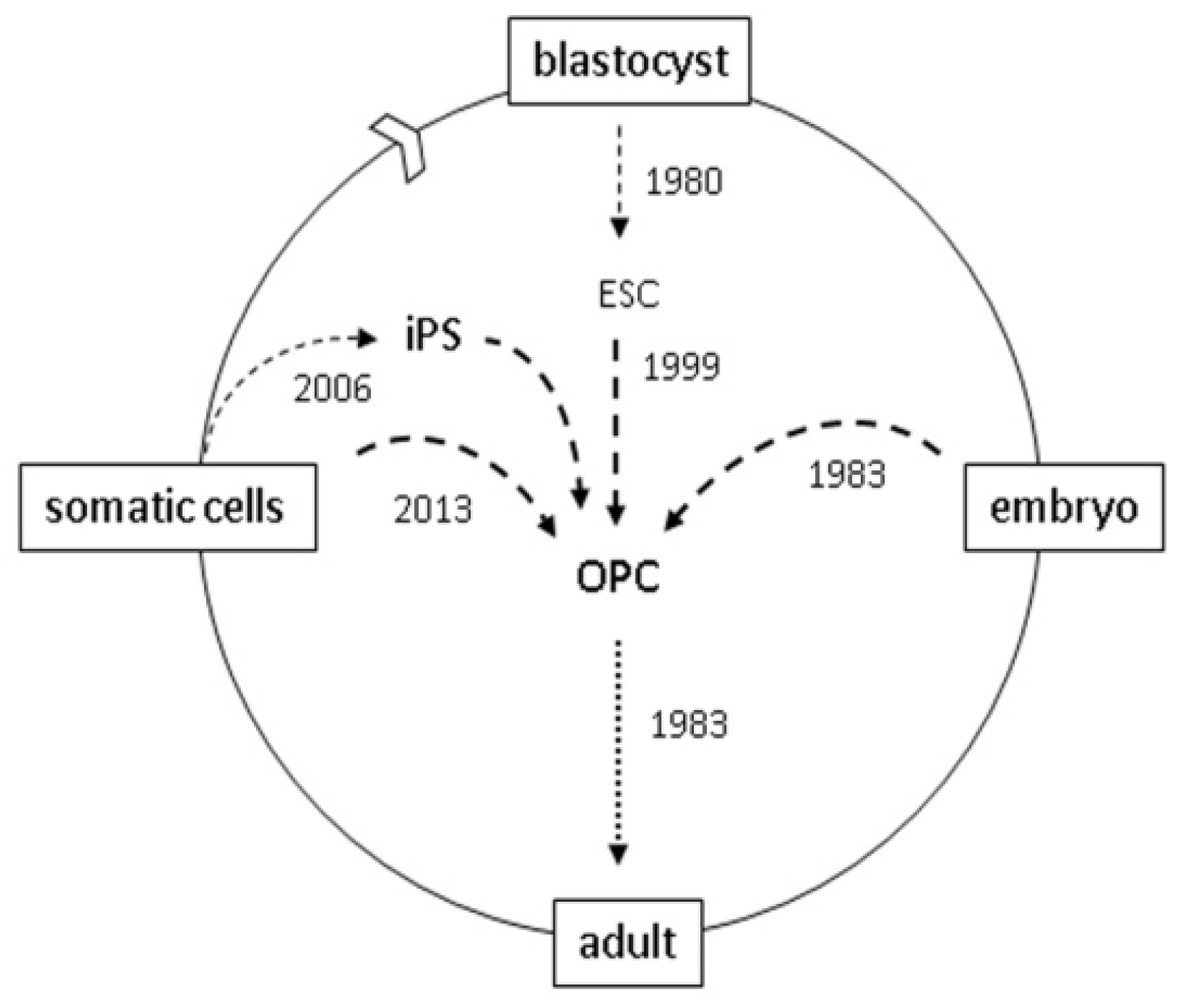
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pre-Clinical Transplants
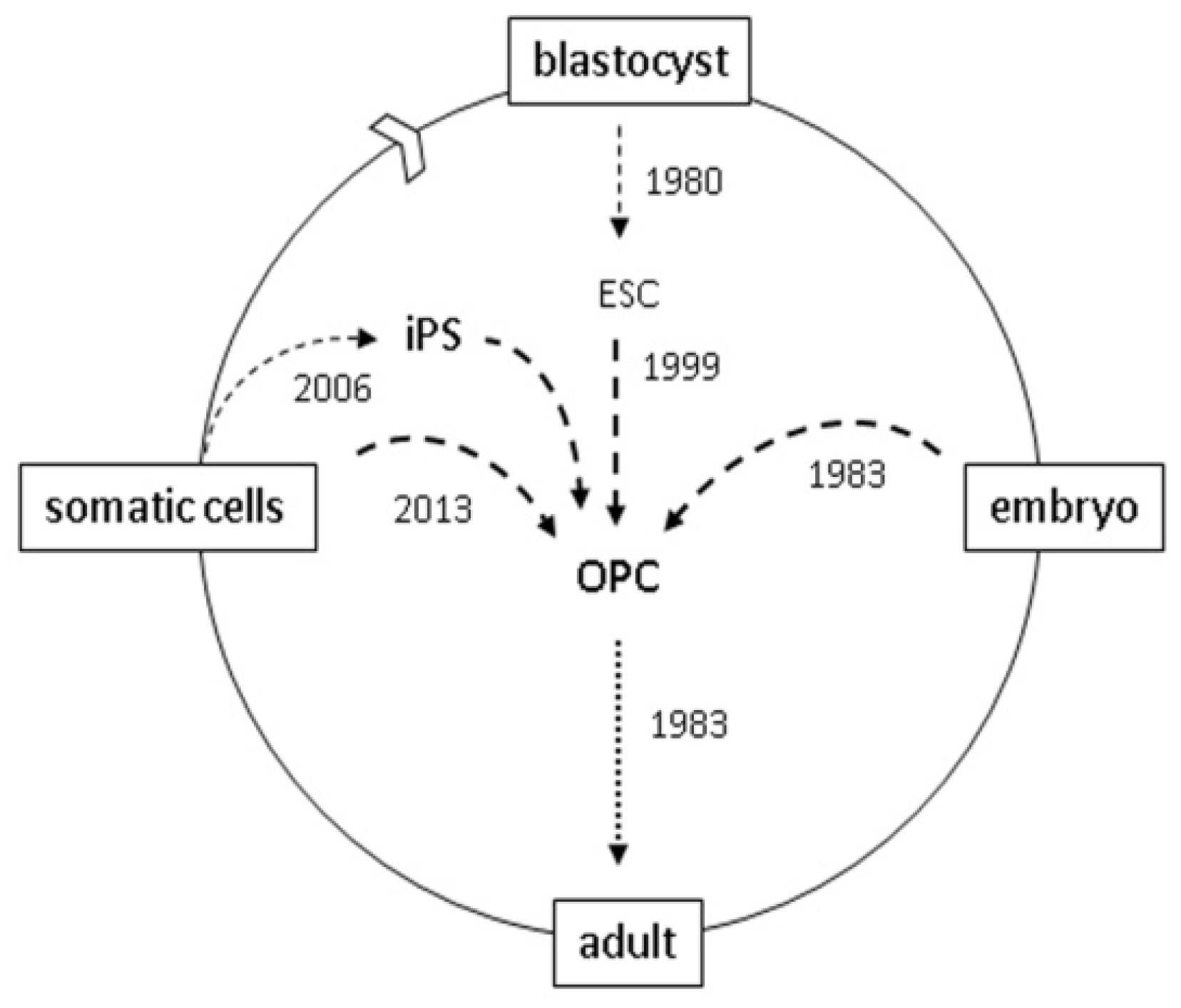
3. Clinical Transplants
4. Reprogramming
5. Direct Reprogramming
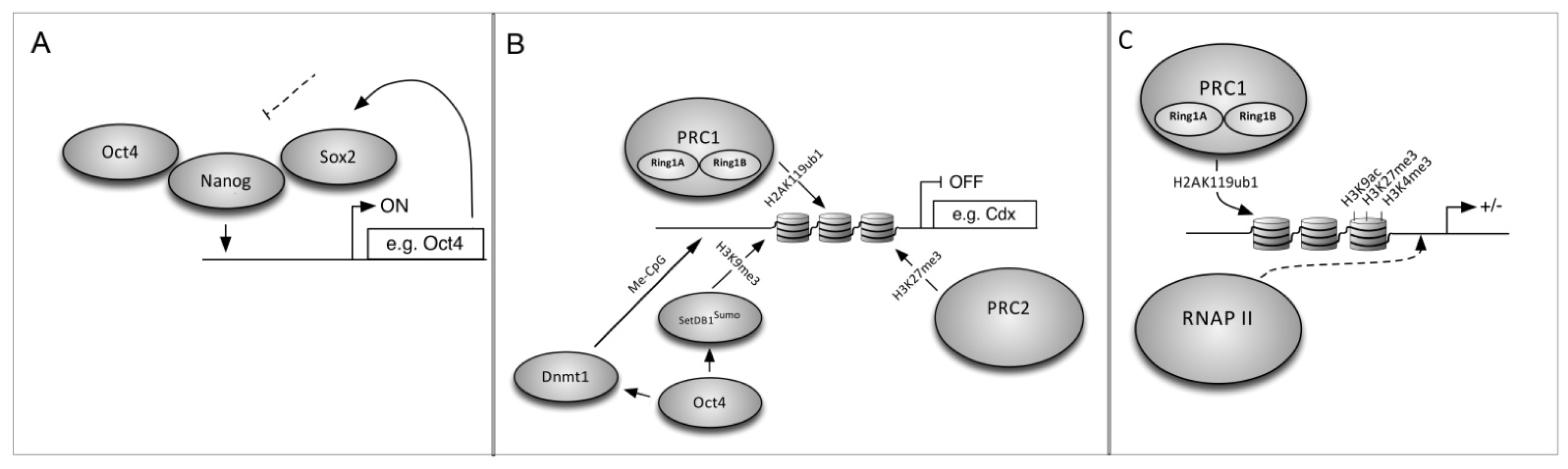
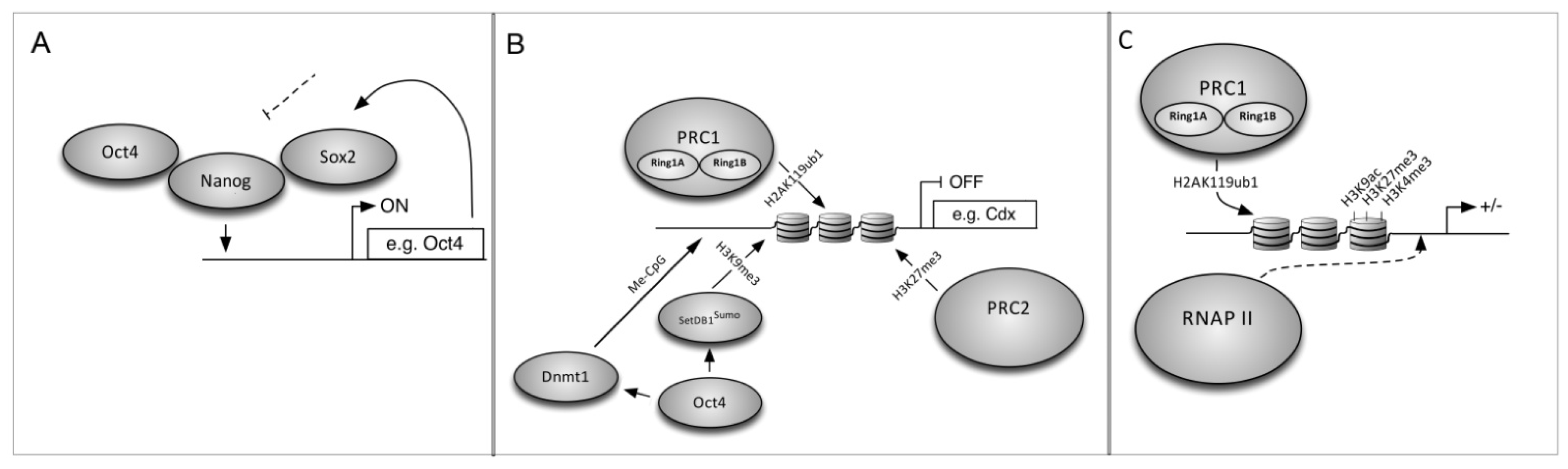
6. The Mechanism of Cell Reprogramming
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Filbin, M.T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003, 4, 703–713. [Google Scholar] [CrossRef]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef]
- Blight, A.R. Miracles and molecules–progress in spinal cord repair. Nat. Neurosci. 2002, 5, 1051–1054. [Google Scholar] [CrossRef]
- Bechmann, I. Failed central nervous system regeneration—A downside of immune privilege? Neuromol. Med. 2005, 7, 217–228. [Google Scholar] [CrossRef]
- Lachapelle, F.; Gumpel, M.; Baulac, M.; Jacque, C.; Duc, P.; Baumann, N. Transplantation of CNS fragments into the brain of shiverer mutant mice: Extensive myelination by implanted oligodendrocytes. I. Immunohistochemical Studies. Dev. Neurosci. 1983, 6, 325–334. [Google Scholar] [CrossRef]
- Bird, T.D.; Farrell, D.F.; Sumi, S.M. Genetic developmental myelin defect in shiverer mouse. Trans. Am. Soc. Neurochem. 1977, 8, 153. [Google Scholar]
- Raff, M.C.; Miller, R.H.; Noble, M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 1983, 303, 390–396. [Google Scholar] [CrossRef]
- Richardson, W.D.; Pringle, N.; Mosley, M.J.; Westermark, B.; Dubois-Dalcq, M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 1988, 53, 309–319. [Google Scholar] [CrossRef]
- McKinnon, R.D.; Matsui, T.; Dubois-Dalcq, M.; Aaronson, S.A. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 1990, 5, 603–614. [Google Scholar] [CrossRef]
- Groves, A.K.; Barnett, S.C.; Franklin, R.J.M.; Crang, A.J.; Mayer, M.; Blakemore, W.F.; Noble, M. Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature 1993, 362, 453–455. [Google Scholar] [CrossRef]
- Warrington, A.E.; Barbarese, E.; Pfeiffer, S.E. Differential myelinogenic capacity of specific developmental stages of the oligodendrocyte lineage upon transplantation into hypomyelinating hosts. J. Neurosci. Res. 1993, 34, 1–13. [Google Scholar] [CrossRef]
- Kuhn, P.L.; Petroulakais, E.; Zazanis, G.A.; McKinnon, R.D. Motor function analysis of myelin mutant mice using a rotarod. Int. J. Dev. Neurosci. 1995, 13, 715–722. [Google Scholar] [CrossRef]
- Kiel, M.E.; Chen, C.P.; Sadowski, D.; McKinnon, R.D. Stem cell-derived therapeutic myelin repair requires 7% cell replacement. Stem Cells 2008, 26, 2229–2236. [Google Scholar] [CrossRef]
- Windrem, M.S.; Schanz, S.J.; Guo, M.; Tian, G.F.; Washco, V.; Stanwood, N.; Rasband, M.; Roy, N.S.; Nedergaard, M.; Havton, L.A.; et al. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2008, 2, 553–565. [Google Scholar] [CrossRef]
- Arriola, A.; Kiel, M.E.; Shi, Y.F.; McKinnon, R.D. Adjunctive MSCs enhance myelin formation by xenogenic oligodendrocyte precursors transplanted in the retina. Cell Res. 2010, 20, 728–731. [Google Scholar] [CrossRef]
- Brustle, O.; Jones, K.N.; Learish, R.D.; Karram, K.; Choudhary, K.; Wiestler, O.D.; Duncan, I.D.; McKay, R.D.G. Embryonic stem cell-derived glial precursors: A source of myelinating transplants. Science 1999, 285, 754–756. [Google Scholar] [CrossRef]
- Keirstead, H.S.; Nistor, G.; Bernal, G.; Totoiu, M.; Cloutier, F.; Sharp, K.; Steward, O. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J. Neurosci. 2005, 25, 4694–4705. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Zuchero, J.B.; Ahlenius, H.; Marro, S.; Ng, Y.H.; Vierbuchen, T.; Hawkins, J.S.; Geissler, R.; Barres, B.A.; Wernig, M. Generation of oligodendroglial cells by direct lineage conversion. Nat. Biotechnol. 2013, 31, 434–439. [Google Scholar] [CrossRef]
- Najm, F.J.; Lager, A.M.; Zaremba, A.; Wyatt, K.; Caprariello, A.V.; Factor, D.C.; Karl, R.T.; Maeda, T.; Miller, R.H.; Tesar, P.J. Transcription factor-mediated reprogramming of fibroblasts to expandable, myelinogenicoligodendrocyte progenitor cells. Nat. Biotechnol. 2013, 31, 426–433. [Google Scholar] [CrossRef]
- Bjorklund, A.; Dunnett, S.B.; Brundin, P.; Stoessl, A.J.; Freed, C.R.; Breeze, R.E.; Levivier, M.; Peschanski, M.; Studer, L.; Barker, R. Neural transplantation for the treatment of Parkinson’s disease. Lancet Neurol. 2003, 2, 437–445. [Google Scholar] [CrossRef]
- Freed, C.R.; Breeze, R.E.; Greene, P.; Fahn, S.; Tsai, W.Y.; Trojanowski, J.Q.; Eidelberg, D. Transplanted dopaminergic neurons: More or less? Nat. Med. 2001, 7, 512–513. [Google Scholar]
- Barberi, T.; Klivenyi, P.; Calingasan, N.Y.; Lee, H.; Kawamata, H.; Loonam, K.; Perrier, A.L.; Bruses, J.; Rubio, M.E.; Topf, N.; et al. Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat. Biotechnol. 2003, 21, 1200–1207. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Totoiu, M.O.; Nistor, G.I.; Lane, T.E.; Keirstead, H.S. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp. Neurol. 2004, 187, 254–265. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Amariglio, N.; Hirshberg, A.; Scheithauer, B.W.; Cohen, Y.; Loewenthal, R.; Trakhtenbrot, L.; Paz, N.; Koren-Michowitz, M.; Waldman, D.; Leider-Trejo, L.; et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 2009, 6, 221–231. [Google Scholar]
- Sadowski, D.; Kiel, M.E.; Apicella, M.; Arriola, A.G.; Chen, C.P.; McKinnon, R.D. Teratogenic potential in cultures optimized for oligodendrocyte development from mouse embryonic stem cells. Stem Cells Dev. 2010, 19, 1343–1353. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Uehlinger, V. “Fertile” intestine nuclei. Nature 1966, 210, 1240–1241. [Google Scholar] [CrossRef]
- Briggs, R.; King, T.J. Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs. Proc. Natl. Acad. Sci. USA 1952, 38, 455–463. [Google Scholar] [CrossRef]
- Daley, G.Q. Cellular alchemy and the golden age of reprogramming. Cell 2012, 151, 1151–1154. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Melton, D.A. Nuclear reprogramming in cells. Science 2008, 322, 1811–1815. [Google Scholar] [CrossRef]
- Tapscott, S.J.; Davis, R.L.; Thayer, M.J.; Cheng, P.F.; Weintraub, H.; Lassar, A.B. MyoD1: A nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 1988, 242, 405–411. [Google Scholar]
- Britten, R.J.; Davidson, E.H. Gene regulation for higher cells: A theory. Science 1969, 165, 349–357. [Google Scholar]
- Yu, J.Y.; Hu, K.J.; Smuga-Otto, K.; Tian, S.L.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef]
- Kim, J.B.; Greber, B.; Arauzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Scholer, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Li, W.L.; Zhou, H.Y.; Wei, W.G.; Ambasudhan, R.; Lin, T.X.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.L.; Ku, M.C.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Lin, T.X.; Ambasudhan, R.; Yuan, X.; Li, W.L.; Hilcove, S.; Abujarour, R.; Lin, X.Y.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef]
- Huangfu, D.W.; Maehr, R.; Guo, W.J.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef]
- Rideout, W.M., III; Hochedlinger, K.; Kyba, M.; Daley, G.Q.; Jaenisch, R. Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 2002, 109, 17–27. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–84. [Google Scholar] [CrossRef]
- Raff, M. Adult stem cell plasticity: Fact or artifact? Annu. Rev. Cell Dev. Biol. 2003, 19, 1–22. [Google Scholar] [CrossRef]
- Woodbury, D.; Schwarz, E.J.; Prockop, D.J.; Black, I.B. Adult rat and human bone marrow stromal cells differentiate into neurons. J. Neurosci. Res. 2000, 61, 364–370. [Google Scholar] [CrossRef]
- Bertani, N.; Malatesta, P.; Volpi, G.; Sonego, P.; Perris, R. Neurogenic potential of human mesenchymal stem cells revisited: analysis by immunostaining, time-lapse video and microarray. J. Cell Sci. 2005, 118, 3925–3936. [Google Scholar] [CrossRef]
- Lagasse, E.; Connors, H.; Al Dhalimy, M.; Reitsma, M.; Dohse, M.; Osborne, L.; Wang, X.; Finegold, M.; Weissman, I.L.; Grompe, M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 2000, 6, 1229–1234. [Google Scholar] [CrossRef]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Wernig, M. Molecular roadblocks for cellular reprogramming. Mol. Cell 2012, 47, 827–838. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Sekiya, S.; Suzuki, A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011, 475, 390–393. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Efe, J.A.; Hilcove, S.; Kim, J.; Zhou, H.; Ouyang, K.; Wang, G.; Chen, J.; Ding, S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 2011, 13, 215–222. [Google Scholar] [CrossRef]
- Szabo, E.; Rampalli, S.; Risueno, R.M.; Schnerch, A.; Mitchell, R.; Fiebig-Comyn, A.; Levadoux-Martin, M.; Bhatia, M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 2010, 468, 521–526. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Bjorklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef]
- Son, E.Y.; Ichida, J.K.; Wainger, B.J.; Toma, J.S.; Rafuse, V.F.; Woolf, C.J.; Eggan, K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell. 2011, 9, 205–218. [Google Scholar] [CrossRef]
- Karow, M.; Sanchez, R.; Schichor, C.; Masserdotti, G.; Ortega, F.; Heinrich, C.; Gascon, S.; Khan, M.A.; Lie, D.C.; Dellavalle, A.; et al. Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 2012, 11, 471–476. [Google Scholar] [CrossRef]
- Yamanaka, S. Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009, 460, 49–52. [Google Scholar] [CrossRef]
- Chang, H.H.; Hemberg, M.; Barahona, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.M.; Ratanasirintrawoot, S.; Park, I.H.; Manos, P.D.; Loh, Y.H.; Huo, H.G.; Miller, J.D.; Hartung, O.; Rho, J.; Ince, T.A.; et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009, 27, 1033–1037. [Google Scholar] [CrossRef]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef]
- Tursun, B.; Patel, T.; Kratsios, P.; Hobert, O. Direct conversion of C. elegans germ cells into specific neuron types. Science 2011, 331, 304–308. [Google Scholar] [CrossRef]
- Simonsson, S.; Gurdon, J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat. Cell Biol. 2004, 6, 984–990. [Google Scholar] [CrossRef]
- Silva, J.; Nichols, J.; Theunissen, T.W.; Guo, G.; van Oosten, A.L.; Barrandon, O.; Wray, J.; Yamanaka, S.; Chambers, I.; Smith, A. Nanog is the gateway to the pluripotent ground state. Cell 2009, 138, 722–737. [Google Scholar] [CrossRef]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef]
- Silva, J.; Smith, A. Capturing pluripotency. Cell 2008, 132, 532–536. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef]
- Niwa, H.; Miyazaki, J.; Smith, A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef]
- Yasuda, S.Y.; Tsuneyoshi, N.; Sumi, T.; Hasegawa, K.; Tada, T.; Nakatsuji, N.; Suemori, H. NANOG maintains self-renewal of primate ES cells in the absence of a feeder layer. Genes Cells 2006, 11, 1115–1123. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Yeap, L.S.; Hayashi, K.; Surani, M.A. ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenetics Chromatin. 2009, 2, 12. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J., III; Voigt, P.; Martin, S.R.; Taylor, W.R.; De, M.V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef]
- Charoensawan, V.; Janga, S.C.; Bulyk, M.L.; Babu, M.M.; Teichmann, S.A. DNA sequence preferences of transcriptional activators correlate more strongly than repressors with nucleosomes. Mol. Cell 2012, 47, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Vierbuchen, T.; Wernig, M. Direct lineage conversions: unnatural but useful? Nat. Biotechnol. 2011, 29, 892–907. [Google Scholar] [CrossRef]
- Smale, S.T. Pioneer factors in embryonic stem cells and differentiation. Curr. Opin. Genet. Dev. 2010, 20, 519–526. [Google Scholar] [CrossRef]
- Huangfu, D.W.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef]
- Rizzino, A. Sox2 and Oct-3/4: A versatile pair of master regulators that orchestrate the self-renewal and pluripotency of embryonic stem cells. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 228–236. [Google Scholar] [CrossRef]
- Kuzmichev, A.N.; Kim, S.K.; D’Alessio, A.C.; Chenoweth, J.G.; Wittko, I.M.; Campanati, L.; McKay, R.D. Sox2 acts through Sox21 to regulate transcription in pluripotent and differentiated cells. Curr. Biol. 2012, 22, 1705–1710. [Google Scholar] [CrossRef]
- Jeong, C.H.; Cho, Y.Y.; Kim, M.O.; Kim, S.H.; Cho, E.J.; Lee, S.Y.; Jeon, Y.J.; Lee, K.Y.; Yao, K.; Keum, Y.S.; et al. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 2010, 28, 2141–2150. [Google Scholar] [CrossRef]
- Tsai, S.Y.; Bouwman, B.A.; Ang, Y.S.; Kim, S.J.; Lee, D.F.; Lemischka, I.R.; Rendl, M. Single transcription factor reprogramming of hair follicle dermal papilla cells to induced pluripotent stem cells. Stem Cells 2011, 29, 964–971. [Google Scholar] [CrossRef]
- Fouse, S.D.; Shen, Y.; Pellegrini, M.; Cole, S.; Meissner, A.; Van Neste, L.; Jaenisch, R.; Fan, G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008, 2, 160–169. [Google Scholar] [CrossRef]
- Taylor, S.M.; Jones, P.A. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell 1979, 17, 771–779. [Google Scholar] [CrossRef]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Arauzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den, B.D.; et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef]
- Yoo, A.S.; Staahl, B.T.; Chen, L.; Crabtree, G.R. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 2009, 460, 642–646. [Google Scholar]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef]
- Wagner, B.K. Grand challenge commentary: Chemical trans differentiation and regenerative medicine. Nat. Chem. Biol. 2010, 6, 877–879. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guarino, A.T.; McKinnon, R.D. Reprogramming Cells for Brain Repair. Brain Sci. 2013, 3, 1215-1228. https://doi.org/10.3390/brainsci3031215
Guarino AT, McKinnon RD. Reprogramming Cells for Brain Repair. Brain Sciences. 2013; 3(3):1215-1228. https://doi.org/10.3390/brainsci3031215
Chicago/Turabian StyleGuarino, Alyx T., and Randall D. McKinnon. 2013. "Reprogramming Cells for Brain Repair" Brain Sciences 3, no. 3: 1215-1228. https://doi.org/10.3390/brainsci3031215