Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination

Abstract
:1. Introduction
2. Myelin Composition and Architecture
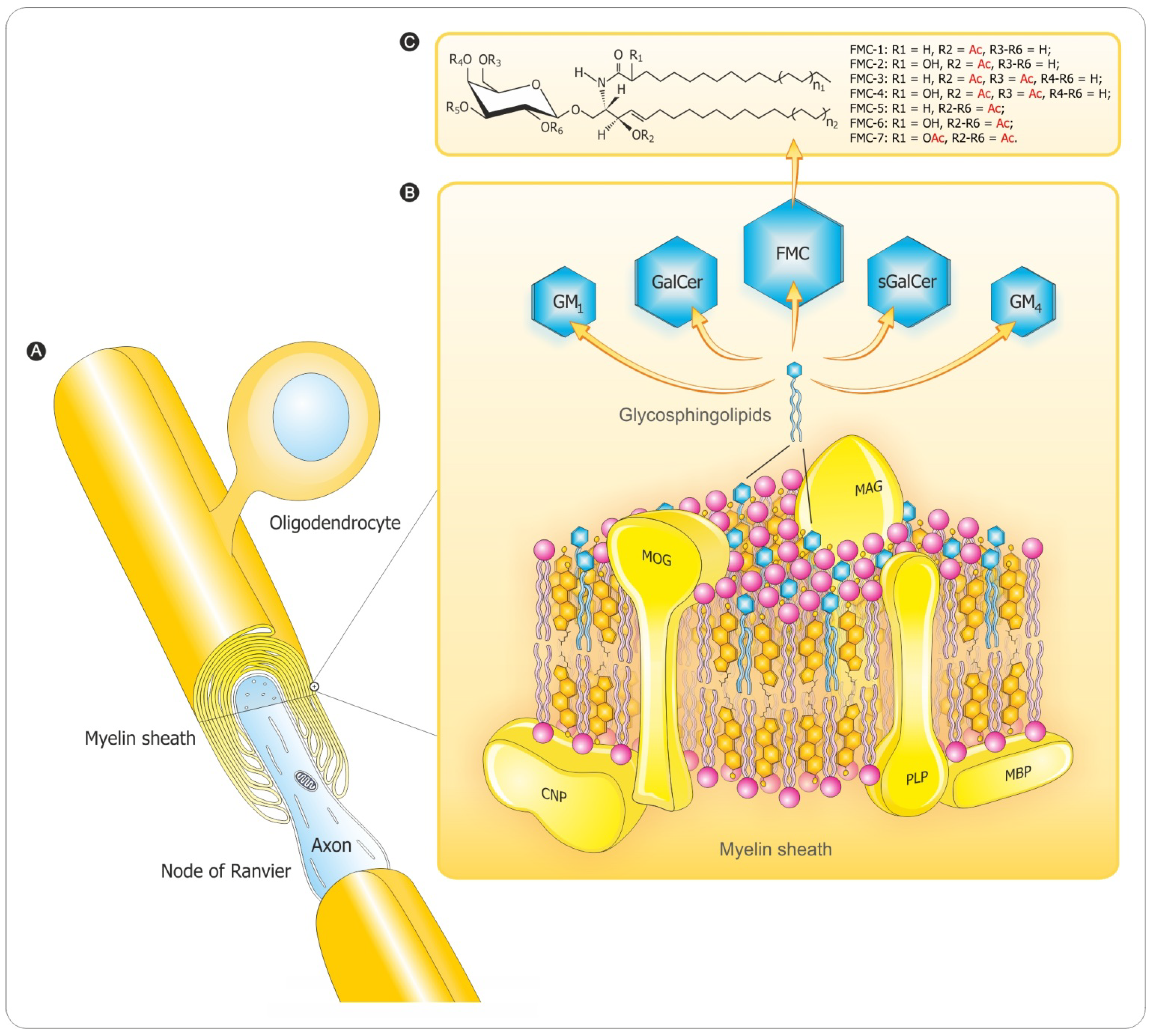
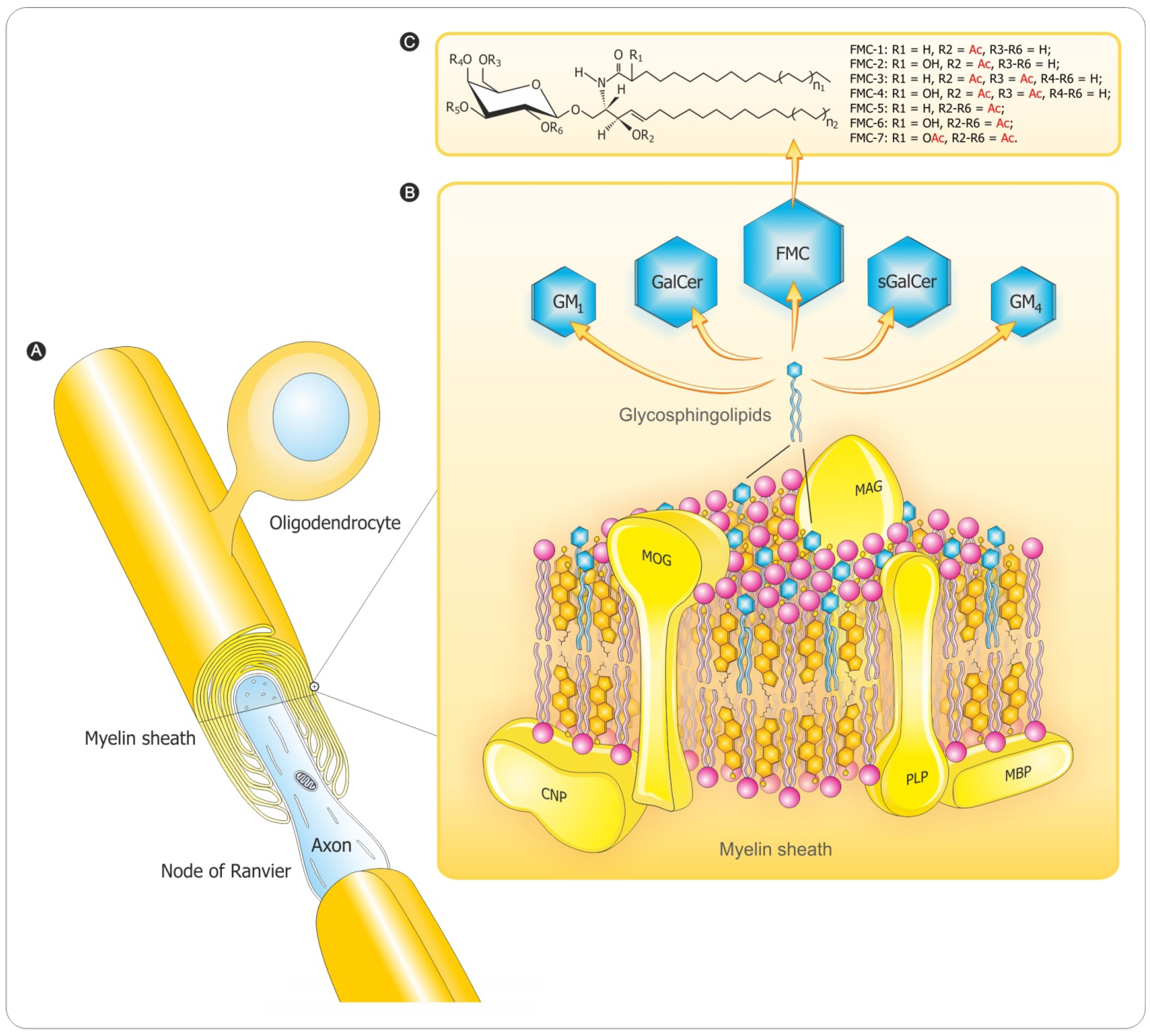
3. Myelination Process
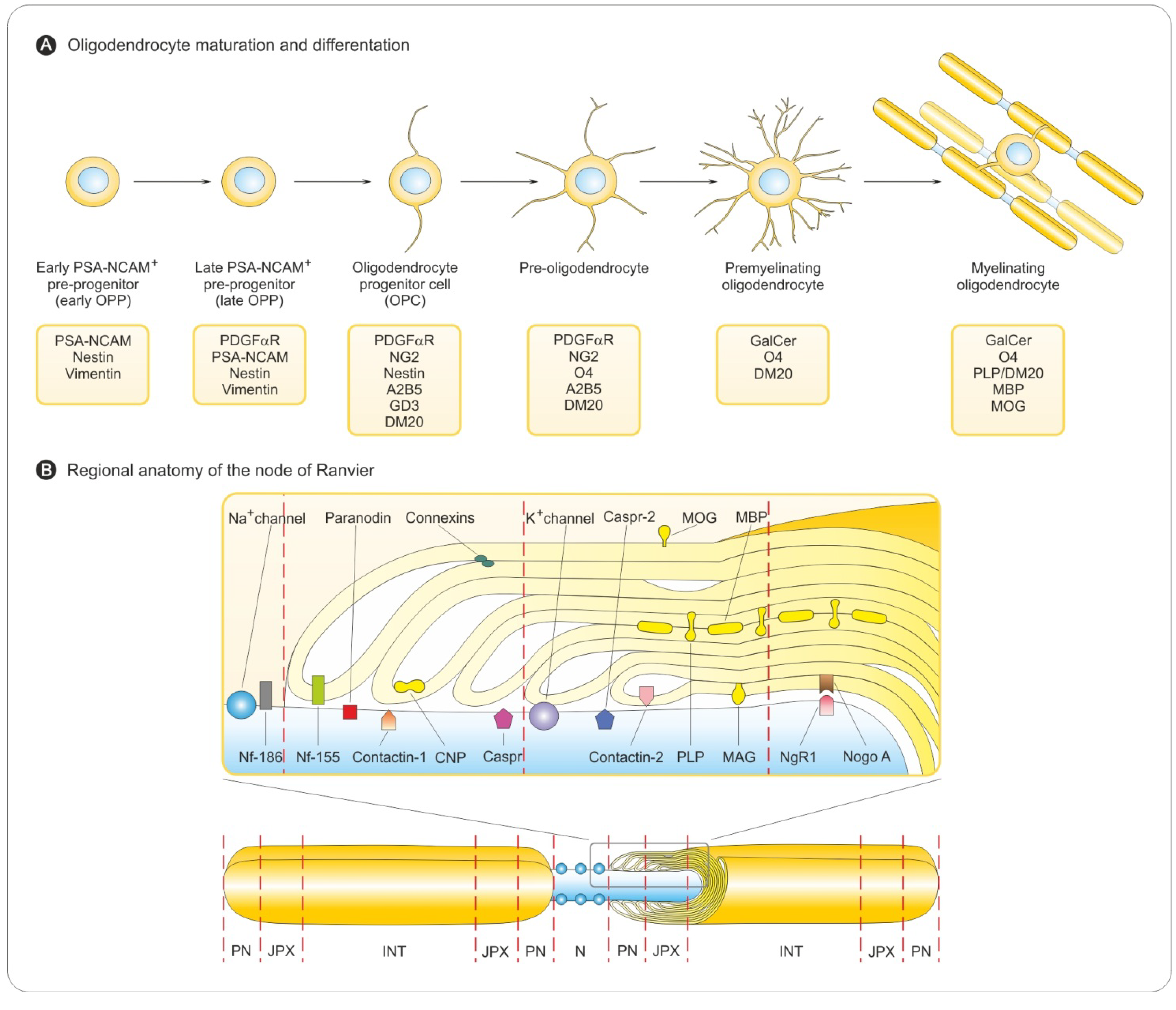
3.1. Oligodendrocyte Differentiation
3.2. Myelin Sheath Organization in the CNS
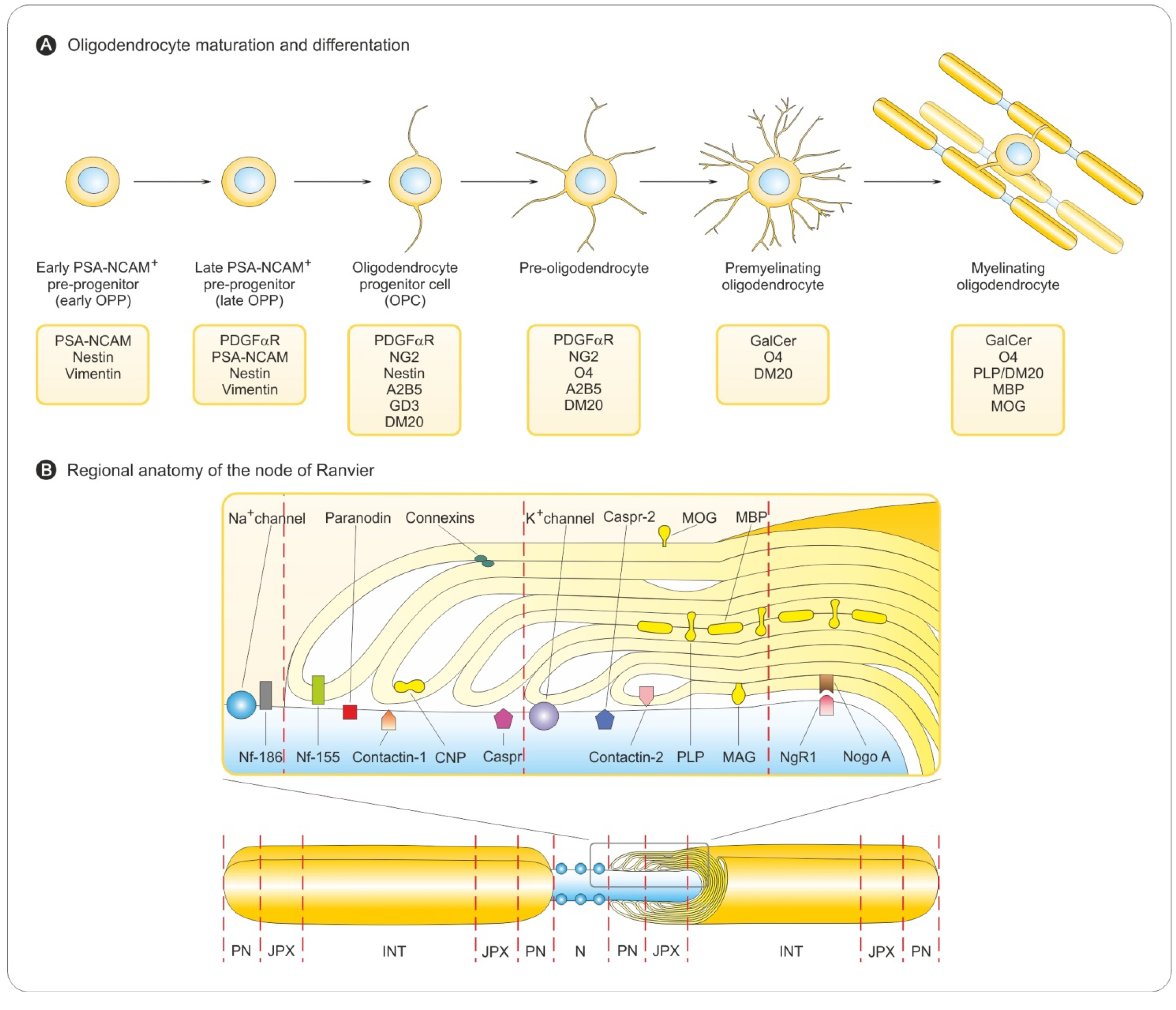
3.3. Myelination in the Central and Peripheral Nervous System
4. Demyelination Process
4.1. Pathology of MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNS | PNS |
|---|---|
| Myelin sheaths are formed by oligodendrocytes. | Myelin sheaths are formed by Schwann cells (the only glial cell type in peripheral nerves). The myelination of PNS axons by Schwann cells is characterized by the sequential appearance of three different types of nerve fibers: (fetal, promyelin, and myelinated nerve fibers). |
| Myelination appears when axonal diameter is >0.2 μm. | Myelination occurs only if axonal diameter is >0.7 μm. |
| A single oligodendrocyte myelinates portions of multiple adjacent axons. | Schwann cells myelinate only one segment of a single axon. |
| Oligodendrocytes lack plasticity. | Schwann cells have remarkable plasticity. |
| Glycoproteins are minor constituents of CNS myelin. | Glycoproteins constitute at least 60% of PNS myelin proteins. |
| Cholesterol required for myelin synthesis is produced by the oligodendrocytes. | Peripheral nerves are hypomyelinated if cholesterol biosynthesis is lacking in Schwann cells [50]. |
| Remyelination requires recruitment and differentiation of progenitor cells into new oligodendrocytes. | Schwann cells can dedifferentiate and assume an immature cell phenotype similar in response to injury. |
| Limited capacity of regeneration of central axons. | Peripheral axons regrow spontaneously after injury in a permissive environment reflecting the intrinsic regenerative capacity of neurons [51]. |
| Remyelination is regulated by axonal signals that differ for oligodendrocytes and Schwann cells [52,53]. | Remyelination is regulated by axonal signals different from those for oligodendrocytes [52,53]. Schwann cells only express a myelinating phenotype when contacting large axons producing threshold levels of neuregulin-1 type III [54]. |
| CNS remyelination can also be achieved by Schwann cells [55] or by immature CNS glia with pluripotent capacity [56]. | PNS demyelination reflects Wallerian degeneration and subsequent regeneration [57]. |
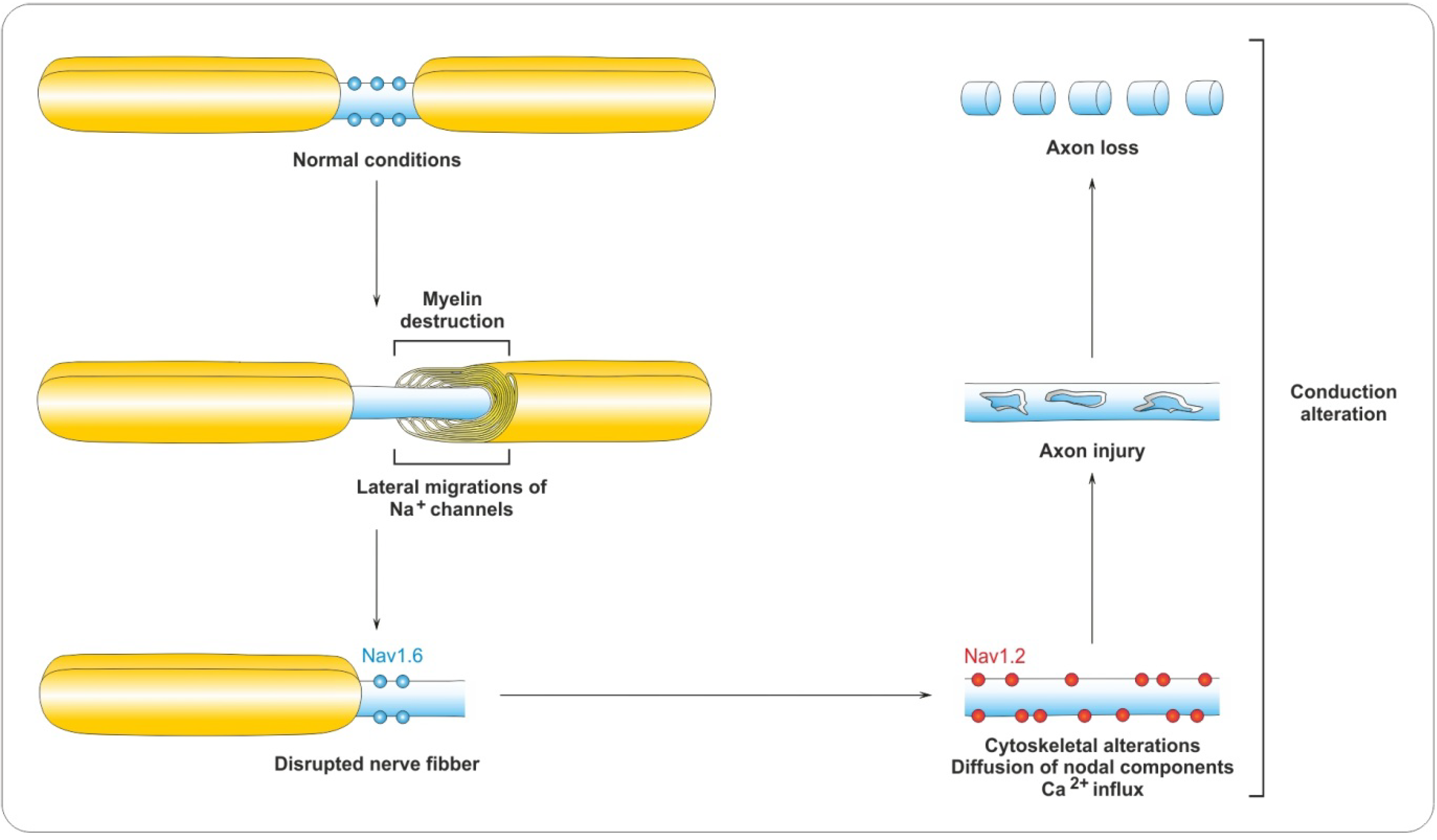
4.2. Myelin Destruction
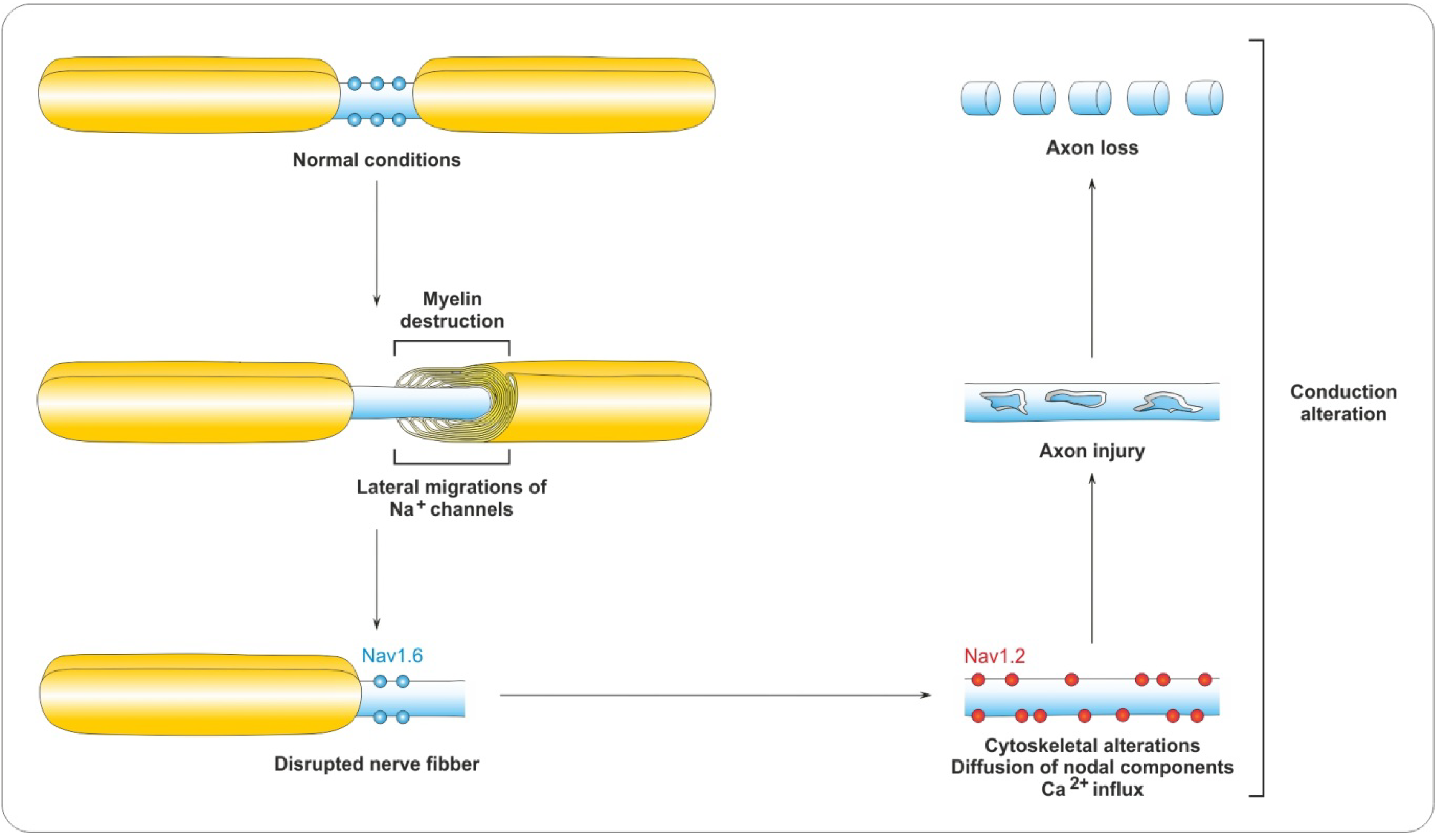
5. Remyelination Process
5.1. Remyelination as a True Regenerative Process
5.2. Role of OPCs in Remyelination
5.3. Restoration of the Myelin Architecture and Impulse Conduction
6. Regulation of Remyelination
6.1. PSA-NCAM
6.2. Notch Signaling
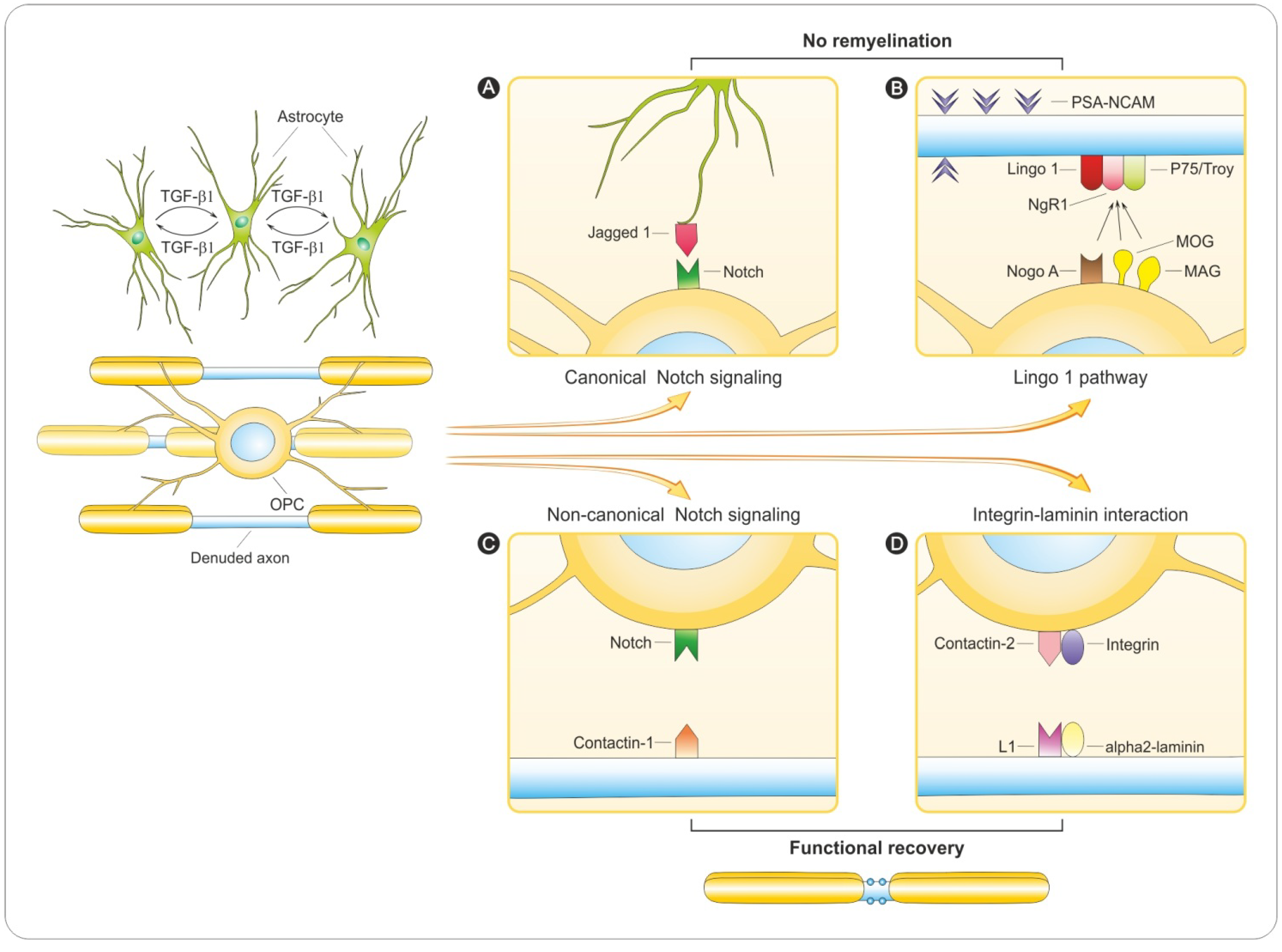
6.3. LINGO-1 Pathway
6.4. Integrin-Laminin Interaction
6.5. Why Does Remyelination Fail?
7. Cytokine-Based Immuno-Intervention
7.1. Inflammation and Repair in MS under Cytokines Control
7.2. Anti-Inflammatory Effect of Calpain Inhibition in MS
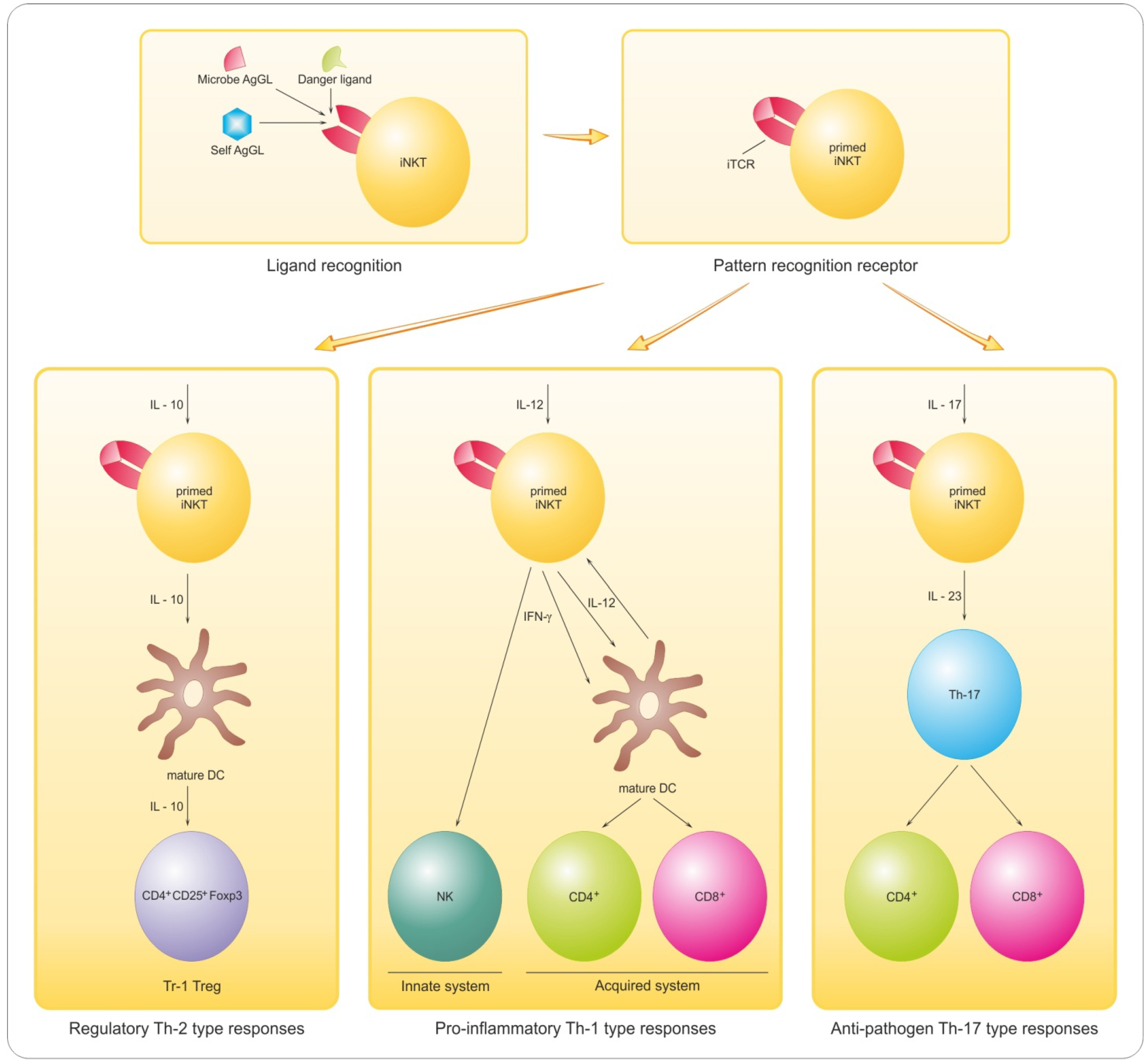
8. Antigen-Based Immune-Intervention
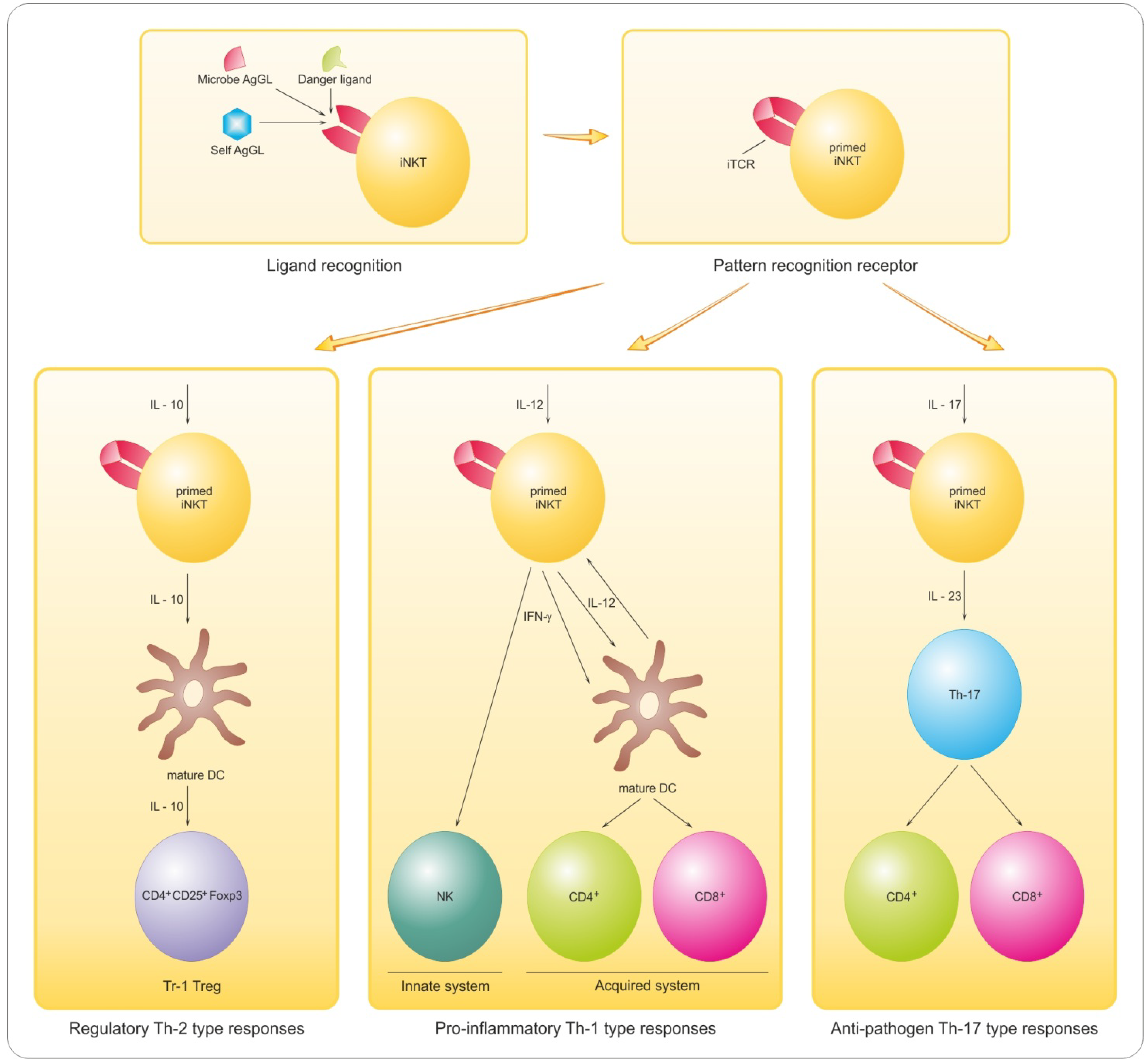
8.1. Implications of Lymphocyte GL Reactivity and Anergy for Myelin Repair
8.2. Promyelinating Antibodies and Potential for Remyelination in MS
8.3. Sphingoid Mediation of Inflammatory Demyelination and Treatment Options
9. Conclusions
Acknowledgments
Conflict of Interest
References
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D. Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 2012, 72, 18–31. [Google Scholar] [CrossRef]
- Franklin, R.J. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef]
- Aharoni, R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun. Rev. 2013, 12, 543–553. [Google Scholar] [CrossRef]
- Rosetti, C.M.; Maggio, B.; Oliveira, R.G. The self-organization of lipids and proteins of myelin at the membrane interface. Molecular factors underlying the microheterogeneity of domain segregation. Biochim. Biophys. Acta 2008, 1778, 1665–1675. [Google Scholar] [CrossRef]
- Norton, W.T.; Autilio, L.A. The lipid composition of purified bovine brain myelin. J. Neurochem. 1966, 13, 213–222. [Google Scholar] [CrossRef]
- Fewster, M.E.; Hirono, H.; Mead, J.F. Lipid composition of myelin in multiple sclerosis. J. Neurol. 1976, 213, 119–131. [Google Scholar] [CrossRef]
- Dasgupta, S.; Levery, S.B.; Hogan, E.L. 3-O-acetyl-sphingosine-series myelin glycolipids: Characterization of novel 3-O-acetyl-sphingosine galactosylceramide. J. Lipid Res. 2002, 43, 751–761. [Google Scholar]
- Bennion, B.; Dasgupta, S.; Hogan, E.L.; Levery, S.B. Characterization of novel myelin components 3-O-acetyl-sphingosine galactosylceramides by electrospray ionization Q-TOF MS and MS/CID-MS of Li+ adducts. J. Mass Spectrom. 2007, 42, 598–620. [Google Scholar] [CrossRef]
- Podbielska, M.; Dasgupta, S.; Levery, S.B.; Tourtellotte, W.W.; Annuk, H.; Moran, A.P.; Hogan, E.L. Novel myelin penta- and hexa-acetyl-galactosyl-ceramides: Structural characterization and immunoreactivity in cerebrospinal fluid. J. Lipid Res. 2010, 51, 1394–1406. [Google Scholar] [CrossRef]
- Yu, R.K.; Ledeen, R.W. Gangliosides of human, bovine, and rabbit plasma. J. Lipid Res. 1972, 13, 680–686. [Google Scholar]
- Xu, L.; Anchordoquy, T.J. Cholesterol domains in cationic lipid/DNA complexes improve transfection. Biochim. Biophys. Acta 2008, 1778, 2177–2181. [Google Scholar] [CrossRef]
- Xu, Y.; Ramu, Y.; Lu, Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature 2008, 451, 826–829. [Google Scholar] [CrossRef]
- Wheeler, D.; Bandaru, V.V.; Calabresi, P.A.; Nath, A.; Haughey, N.J. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 2008, 131, 3092–3102. [Google Scholar] [CrossRef]
- Podbielska, M.; Hogan, E.L. Molecular and immunogenic features of myelin lipids: Incitants or modulators of multiple sclerosis? Mult. Scler. 2009, 15, 1011–1029. [Google Scholar] [CrossRef]
- Podbielska, M.; Levery, S.B.; Hogan, E.L. The structural and functional role of myelin fast-migrating cerebrosides: Pathological importance in multiple sclerosis. Clin. Lipidol. 2011, 6, 159–179. [Google Scholar] [CrossRef]
- Magavi, S.S.; Leavitt, B.R.; Macklis, J.D. Induction of neurogenesis in the neocortex of adult mice. Nature 2000, 405, 951–955. [Google Scholar] [CrossRef]
- Richardson, W.D.; Kessaris, N.; Pringle, N. Oligodendrocyte wars. Nat. Rev. Neurosci. 2006, 7, 11–18. [Google Scholar] [CrossRef]
- Noll, E.; Miller, R.H. Oligodendrocyte precursors originate at the ventral ventricular zone dorsal to the ventral midline region in the embryonic rat spinal cord. Development 1993, 118, 563–573. [Google Scholar]
- Pringle, N.P.; Guthrie, S.; Lumsden, A.; Richardson, W.D. Dorsal spinal cord neuroepithelium generates astrocytes but not oligodendrocytes. Neuron 1998, 20, 883–893. [Google Scholar] [CrossRef]
- Fu, H.; Qi, Y.; Tan, M.; Cai, J.; Takebayashi, H.; Nakafuku, M.; Richardson, W.; Qiu, M. Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development 2002, 129, 681–693. [Google Scholar]
- Cai, J.; Qi, Y.; Hu, X.; Tan, M.; Liu, Z.; Zhang, J.; Li, Q.; Sander, M.; Qiu, M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron 2005, 45, 41–53. [Google Scholar] [CrossRef]
- Vallstedt, A.; Klos, J.M.; Ericson, J. Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron 2005, 45, 55–67. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Mekki-Dauriac, S.; Agius, E.; Kan, P.; Cochard, P. Bone morphogenetic proteins negatively control oligodendrocyte precursor specification in the chick spinal cord. Development 2002, 129, 5117–5130. [Google Scholar]
- Noble, M.; Fok-Seang, J.; Wolswijk, G.; Wren, D. Development and regeneration in the central nervous system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 327, 127–143. [Google Scholar] [CrossRef]
- Fok-Seang, J.; Miller, R.H. Distribution and differentiation of A2B5+ glial precursors in the developing rat spinal cord. J. Neurosci. Res. 1994, 37, 219–235. [Google Scholar] [CrossRef]
- Richardson, R.M.; Holloway, K.L.; Bullock, M.R.; Broaddus, W.C.; Fillmore, H.L. Isolation of neuronal progenitor cells from the adult human neocortex. Acta Neurochir. 2006, 148, 773–777. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar]
- Miller, R.H. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 2002, 67, 451–467. [Google Scholar] [CrossRef]
- Nishiyama, A. NG2 cells in the brain: A novel glial cell population. Hum. Cell 2001, 14, 77–82. [Google Scholar]
- Dawson, M.R.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Fancy, S.P.; Chan, J.R.; Baranzini, S.E.; Franklin, R.J.; Rowitch, D.H. Myelin regeneration: A recapitulation of development? Annu. Rev. Neurosci. 2011, 34, 21–43. [Google Scholar] [CrossRef]
- Huang, J.K.; Franklin, R.J. Regenerative medicine in multiple sclerosis: Identifying pharmacological targets of adult neural stem cell differentiation. Neurochem. Int. 2011, 59, 329–332. [Google Scholar]
- Pfeiffer, S.E.; Warrington, A.E.; Bansal, R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993, 3, 191–197. [Google Scholar] [CrossRef]
- Lajtha, A.; Toth, J.; Fujimoto, K.; Agrawal, H.C. Turnover of myelin proteins in mouse brain in vivo. Biochem. J. 1977, 164, 323–329. [Google Scholar]
- LeBaron, F.N.; Sanyal, S.; Jungalwala, F.B. Turnover rate of molecular species of sphingomyelin in rat brain. Neurochem. Res. 1981, 6, 1081–1089. [Google Scholar] [CrossRef]
- Baba, H.; Akita, H.; Ishibashi, T.; Inoue, Y.; Nakahira, K.; Ikenaka, K. Completion of myelin compaction, but not the attachment of oligodendroglial processes triggers K+ channel clustering. J. Neurosci. Res. 1999, 58, 752–764. [Google Scholar] [CrossRef]
- Simons, M.; Trajkovic, K. Neuron-glia communication in the control of oligodendrocyte function and myelin biogenesis. J. Cell Sci. 2006, 119, 4381–4389. [Google Scholar] [CrossRef]
- Ishibashi, T.; Dakin, K.A.; Stevens, B.; Lee, P.R.; Kozlov, S.V.; Stewart, C.L.; Fields, R.D. Astrocytes promote myelination in response to electrical impulses. Neuron 2006, 49, 823–832. [Google Scholar] [CrossRef]
- Piaton, G.; Gould, R.M.; Lubetzki, C. Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J. Neurochem. 2010, 114, 1243–1260. [Google Scholar]
- Foster, R.E.; Connors, B.W.; Waxman, S.G. Rat optic nerve: Electrophysiological, pharmacological and anatomical studies during development. Brain Res. 1982, 255, 371–386. [Google Scholar]
- Voyvodic, J.T. Target size regulates calibre and myelination of sympathetic axons. Nature 1989, 342, 430–433. [Google Scholar] [CrossRef]
- Friede, R.L. Control of myelin formation by axon caliber (with a model of the control mechanism). J. Comp. Neurol. 1972, 144, 233–252. [Google Scholar] [CrossRef]
- Garbay, B.; Heape, A.M.; Sargueil, F.; Cassagne, C. Myelin synthesis in the peripheral nervous system. Prog. Neurobiol. 2000, 61, 267–304. [Google Scholar] [CrossRef]
- Saher, G.; Quintes, S.; Mobius, W.; Wehr, M.C.; Kramer-Albers, E.M.; Brugger, B.; Nave, K.A. Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J. Neurosci. 2009, 29, 6094–6104. [Google Scholar] [CrossRef]
- Chen, Z.L.; Yu, W.M.; Strickland, S. Peripheral regeneration. Annu. Rev. Neurosci. 2007, 30, 209–233. [Google Scholar] [CrossRef]
- Chan, J.R.; Watkins, T.A.; Cosgaya, J.M.; Zhang, C.; Chen, L.; Reichardt, L.F.; Shooter, E.M.; Barres, B.A. NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron 2004, 43, 183–191. [Google Scholar] [CrossRef]
- Brinkmann, B.G.; Agarwal, A.; Sereda, M.W.; Garratt, A.N.; Muller, T.; Wende, H.; Stassart, R.M.; Nawaz, S.; Humml, C.; Velanac, V.; et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 2008, 59, 581–595. [Google Scholar] [CrossRef]
- Michailov, G.V.; Sereda, M.W.; Brinkmann, B.G.; Fischer, T.M.; Haug, B.; Birchmeier, C.; Role, L.; Lai, C.; Schwab, M.H.; Nave, K.A. Axonal neuregulin-1 regulates myelin sheath thickness. Science 2004, 304, 700–703. [Google Scholar] [CrossRef]
- Black, J.A.; Waxman, S.G.; Smith, K.J. Remyelination of dorsal column axons by endogenous Schwann cells restores the normal pattern of Nav1.6 and Kv1.2 at nodes of Ranvier. Brain 2006, 129, 1319–1329. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflamm. 2011, 8, 109. [Google Scholar] [CrossRef]
- Srivastava, R.; Aslam, M.; Kalluri, S.R.; Schirmer, L.; Buck, D.; Tackenberg, B.; Rothhammer, V.; Chan, A.; Gold, R.; Berthele, A.; et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N. Engl. J. Med. 2012, 367, 115–123. [Google Scholar] [CrossRef]
- Lassmann, H. Mechanisms of inflammation induced tissue injury in multiple sclerosis. J. Neurol. Sci. 2008, 274, 45–47. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef]
- Becher, B.; Bechmann, I.; Greter, M. Antigen presentation in autoimmunity and CNS inflammation: How T lymphocytes recognize the brain. J. Mol. Med. 2006, 84, 532–543. [Google Scholar] [CrossRef]
- Mukherjee, S.; Soe, T.T.; Maxfield, F.R. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Biol. 1999, 144, 1271–1284. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maxfield, F.R. Role of membrane organization and membrane domains in endocytic lipid trafficking. Traffic 2000, 1, 203–211. [Google Scholar] [CrossRef]
- Sugita, M.; Porcelli, S.A.; Brenner, M.B. Assembly and retention of CD1b heavy chains in the endoplasmic reticulum. J. Immunol. 1997, 159, 2358–2365. [Google Scholar]
- Gadola, S.D.; Zaccai, N.R.; Harlos, K.; Shepherd, D.; Castro-Palomino, J.C.; Ritter, G.; Schmidt, R.R.; Jones, E.Y.; Cerundolo, V. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat. Immunol. 2002, 3, 721–726. [Google Scholar] [CrossRef]
- Batuwangala, T.; Shepherd, D.; Gadola, S.D.; Gibson, K.J.; Zaccai, N.R.; Fersht, A.R.; Besra, G.S.; Cerundolo, V.; Jones, E.Y. The crystal structure of human CD1b with a bound bacterial glycolipid. J. Immunol. 2004, 172, 2382–2388. [Google Scholar]
- Moody, D.B.; Ulrichs, T.; Muhlecker, W.; Young, D.C.; Gurcha, S.S.; Grant, E.; Rosat, J.P.; Brenner, M.B.; Costello, C.E.; Besra, G.S.; et al. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature 2000, 404, 884–888. [Google Scholar] [CrossRef]
- Shamshiev, A.; Gober, H.J.; Donda, A.; Mazorra, Z.; Mori, L.; de Libero, G. Presentation of the same glycolipid by different CD1 molecules. J. Exp. Med. 2002, 195, 1013–1021. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef]
- Alt, C.; Laschinger, M.; Engelhardt, B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2002, 32, 2133–2144. [Google Scholar] [CrossRef]
- Columba-Cabezas, S.; Serafini, B.; Ambrosini, E.; Aloisi, F. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: Implications for the maintenance of chronic neuroinflammation. Brain Pathol. 2003, 13, 38–51. [Google Scholar]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Racke, M.K.; Scott, D.E.; Quigley, L.; Gray, G.S.; Abe, R.; June, C.H.; Perrin, P.J. Distinct roles for B7-1 (CD-80) and B7-2 (CD-86) in the initiation of experimental allergic encephalomyelitis. J. Clin. Investig. 1995, 96, 2195–2203. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Wegmann, K.W.; Funatake, C.; Whitham, R.H. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J. Immunol. 1999, 162, 1818–1826. [Google Scholar]
- Wolswijk, G.; Balesar, R. Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain 2003, 126, 1638–1649. [Google Scholar] [CrossRef]
- Coman, I.; Aigrot, M.S.; Seilhean, D.; Reynolds, R.; Girault, J.A.; Zalc, B.; Lubetzki, C. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 2006, 129, 3186–3195. [Google Scholar] [CrossRef]
- Howell, O.W.; Palser, A.; Polito, A.; Melrose, S.; Zonta, B.; Scheiermann, C.; Vora, A.J.; Brophy, P.J.; Reynolds, R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 2006, 129, 3173–3185. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Vickers, J.C.; King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Staal, J.A.; McCormack, G.H.; Blizzard, C.A.; Musgrove, R.E.; Mitew, S.; Liu, Y.; et al. Axonopathy and cytoskeletal disruption in degenerative diseases of the central nervous system. Brain Res. Bull. 2009, 80, 217–223. [Google Scholar] [CrossRef]
- Aharoni, R.; Arnon, R.; Eilam, R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J. Neurosci. 2005, 25, 8217–8228. [Google Scholar] [CrossRef]
- Liebetanz, D.; Merkler, D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp. Neurol. 2006, 202, 217–224. [Google Scholar] [CrossRef]
- Smith, P.M.; Jeffery, N.D. Histological and ultrastructural analysis of white matter damage after naturally-occurring spinal cord injury. Brain Pathol. 2006, 16, 99–109. [Google Scholar] [CrossRef]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef]
- Gupta, R.; Rowshan, K.; Chao, T.; Mozaffar, T.; Steward, O. Chronic nerve compression induces local demyelination and remyelination in a rat model of carpal tunnel syndrome. Exp. Neurol. 2004, 187, 500–508. [Google Scholar] [CrossRef]
- Raine, C.S.; Wu, E. Multiple sclerosis: Remyelination in acute lesions. J. Neuropathol. Exp. Neurol. 1993, 52, 199–204. [Google Scholar] [CrossRef]
- Hagemeier, K.; Bruck, W.; Kuhlmann, T. Multiple sclerosis—Remyelination failure as a cause of disease progression. Histol. Histopathol. 2012, 27, 277–287. [Google Scholar]
- Nakahara, J.; Kanekura, K.; Nawa, M.; Aiso, S.; Suzuki, N. Abnormal expression of TIP30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J. Clin. Invest. 2009, 119, 169–181. [Google Scholar]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef]
- Prineas, J.W.; Connell, F. Remyelination in multiple sclerosis. Ann. Neurol. 1979, 5, 22–31. [Google Scholar] [CrossRef]
- Redford, E.J.; Smith, K.J.; Gregson, N.A.; Davies, M.; Hughes, P.; Gearing, A.J.; Miller, K.; Hughes, R.A. A combined inhibitor of matrix metalloproteinase activity and tumour necrosis factor-α processing attenuates experimental autoimmune neuritis. Brain 1997, 120, 1895–1905. [Google Scholar] [CrossRef]
- Byravan, S.; Foster, L.M.; Phan, T.; Verity, A.N.; Campagnoni, A.T. Murine oligodendroglial cells express nerve growth factor. Proc. Natl. Acad. Sci. USA 1994, 91, 8812–8816. [Google Scholar] [CrossRef]
- Strelau, J.; Unsicker, K. GDNF family members and their receptors: Expression and functions in two oligodendroglial cell lines representing distinct stages of oligodendroglial development. Glia 1999, 26, 291–301. [Google Scholar] [CrossRef]
- Wilkins, A.; Chandran, S.; Compston, A. A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia 2001, 36, 48–57. [Google Scholar] [CrossRef]
- Nishiyama, A.; Lin, X.H.; Giese, N.; Heldin, C.H.; Stallcup, W.B. Co-localization of NG2 proteoglycan and PDGF α-receptor on O2A progenitor cells in the developing rat brain. J. Neurosci. Res. 1996, 43, 299–314. [Google Scholar] [CrossRef]
- Pringle, N.P.; Mudhar, H.S.; Collarini, E.J.; Richardson, W.D. PDGF receptors in the rat CNS: During late neurogenesis, PDGF α-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development 1992, 115, 535–551. [Google Scholar]
- Glezer, I.; Lapointe, A.; Rivest, S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J. 2006, 20, 750–752. [Google Scholar]
- Rhodes, K.E.; Raivich, G.; Fawcett, J.W. The injury response of oligodendrocyte precursor cells is induced by platelets, macrophages and inflammation-associated cytokines. Neuroscience 2006, 140, 87–100. [Google Scholar] [CrossRef]
- Fancy, S.P.; Zhao, C.; Franklin, R.J. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef]
- Smith, K.J.; Blakemore, W.F.; McDonald, W.I. Central remyelination restores secure conduction. Nature 1979, 280, 395–396. [Google Scholar] [CrossRef]
- Oluich, L.J.; Stratton, J.A.; Xing, Y.L.; Ng, S.W.; Cate, H.S.; Sah, P.; Windels, F.; Kilpatrick, T.J.; Merson, T.D. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J. Neurosci. 2012, 32, 8317–8330. [Google Scholar] [CrossRef]
- Gledhill, R.F.; Harrison, B.M.; McDonald, W.I. Pattern of remyelination in the CNS. Nature 1973, 244, 443–444. [Google Scholar] [CrossRef]
- Blakemore, W.F. Pattern of remyelination in the CNS. Nature 1974, 249, 577–578. [Google Scholar] [CrossRef]
- Ludwin, S.K.; Maitland, M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J. Neurol. Sci. 1984, 64, 193–198. [Google Scholar] [CrossRef]
- Ghatak, N.R.; Leshner, R.T.; Price, A.C.; Felton, W.L. 3rd Remyelination in the human central nervous system. J. Neuropathol. Exp. Neurol. 1989, 48, 507–518. [Google Scholar] [CrossRef]
- Dupree, J.L.; Mason, J.L.; Marcus, J.R.; Stull, M.; Levinson, R.; Matsushima, G.K.; Popko, B. Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biol. 2004, 1, 179–192. [Google Scholar]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef]
- Charles, P.; Reynolds, R.; Seilhean, D.; Rougon, G.; Aigrot, M.S.; Niezgoda, A.; Zalc, B.; Lubetzki, C. Re-expression of PSA-NCAM by demyelinated axons: An inhibitor of remyelination in multiple sclerosis? Brain 2002, 125, 1972–1979. [Google Scholar] [CrossRef]
- Charles, P.; Hernandez, M.P.; Stankoff, B.; Aigrot, M.S.; Colin, C.; Rougon, G.; Zalc, B.; Lubetzki, C. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 7585–7590. [Google Scholar] [CrossRef]
- Jakovcevski, I.; Mo, Z.; Zecevic, N. Down-regulation of the axonal polysialic acid-neural cell adhesion molecule expression coincides with the onset of myelination in the human fetal forebrain. Neuroscience 2007, 149, 328–337. [Google Scholar] [CrossRef]
- Fewou, S.N.; Ramakrishnan, H.; Bussow, H.; Gieselmann, V.; Eckhardt, M. Down-regulation of polysialic acid is required for efficient myelin formation. J. Biol. Chem. 2007, 282, 16700–16711. [Google Scholar] [CrossRef]
- Franceschini, I.; Vitry, S.; Padilla, F.; Casanova, P.; Tham, T.N.; Fukuda, M.; Rougon, G.; Durbec, P.; Dubois-Dalcq, M. Migrating and myelinating potential of neural precursors engineered to overexpress PSA-NCAM. Mol. Cell. Neurosci. 2004, 27, 151–162. [Google Scholar] [CrossRef]
- Zhang, H.; Vutskits, L.; Calaora, V.; Durbec, P.; Kiss, J.Z. A role for the polysialic acid-neural cell adhesion molecule in PDGF-induced chemotaxis of oligodendrocyte precursor cells. J. Cell Sci. 2004, 117, 93–103. [Google Scholar] [CrossRef]
- Taveggia, C.; Feltri, M.L.; Wrabetz, L. Signals to promote myelin formation and repair. Nat. Rev. Neurol. 2010, 6, 276–287. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Selmaj, K. Notch: A new player in MS mechanisms. J. Neuroimmunol. 2010, 218, 3–11. [Google Scholar] [CrossRef]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef]
- Stidworthy, M.F.; Genoud, S.; Li, W.W.; Leone, D.P.; Mantei, N.; Suter, U.; Franklin, R.J. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain 2004, 127, 1928–1941. [Google Scholar] [CrossRef]
- Zhang, Y.; Argaw, A.T.; Gurfein, B.T.; Zameer, A.; Snyder, B.J.; Ge, C.; Lu, Q.R.; Rowitch, D.H.; Raine, C.S.; Brosnan, C.F.; et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl. Acad. Sci. USA 2009, 106, 19162–19167. [Google Scholar] [CrossRef]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef]
- Peress, N.S.; Perillo, E.; Seidman, R.J. Glial transforming growth factor (TGF)-beta isotypes in multiple sclerosis: Differential glial expression of TGF-β 1, 2 and 3 isotypes in multiple sclerosis. J. Neuroimmunol. 1996, 71, 115–123. [Google Scholar] [CrossRef]
- Hu, Q.D.; Ang, B.T.; Karsak, M.; Hu, W.P.; Cui, X.Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.K.; et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef]
- Cui, X.Y.; Hu, Q.D.; Tekaya, M.; Shimoda, Y.; Ang, B.T.; Nie, D.Y.; Sun, L.; Hu, W.P.; Karsak, M.; Duka, T.; et al. NB-3/Notch1 pathway via Deltex1 promotes neural progenitor cell differentiation into oligodendrocytes. J. Biol. Chem. 2004, 279, 25858–25865. [Google Scholar] [CrossRef]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kumpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar]
- Liu, B.P.; Fournier, A.; GrandPre, T.; Strittmatter, S.M. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 2002, 297, 1190–1193. [Google Scholar] [CrossRef]
- Wang, K.C.; Koprivica, V.; Kim, J.A.; Sivasankaran, R.; Guo, Y.; Neve, R.L.; He, Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 2002, 417, 941–944. [Google Scholar] [CrossRef]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000, 403, 434–439. [Google Scholar] [CrossRef]
- Moreau-Fauvarque, C.; Kumanogoh, A.; Camand, E.; Jaillard, C.; Barbin, G.; Boquet, I.; Love, C.; Jones, E.Y.; Kikutani, H.; Lubetzki, C.; et al. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J. Neurosci. 2003, 23, 9229–9239. [Google Scholar]
- Benson, M.D.; Romero, M.I.; Lush, M.E.; Lu, Q.R.; Henkemeyer, M.; Parada, L.F. Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc. Natl. Acad. Sci. USA 2005, 102, 10694–10699. [Google Scholar]
- Fournier, A.E.; GrandPre, T.; Strittmatter, S.M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001, 409, 341–346. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Shao, Z.; Thill, G.; Ji, B.; Relton, J.; Levesque, M.; Allaire, N.; Perrin, S.; Sands, B.; et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 2004, 7, 221–228. [Google Scholar] [CrossRef]
- Mi, S.; Miller, R.H.; Lee, X.; Scott, M.L.; Shulag-Morskaya, S.; Shao, Z.; Chang, J.; Thill, G.; Levesque, M.; Zhang, M.; et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751. [Google Scholar] [CrossRef]
- Mi, S. Troy/Taj and its role in CNS axon regeneration. Cytokine Growth Factor Rev. 2008, 19, 245–251. [Google Scholar] [CrossRef]
- Laursen, L.S.; Chan, C.W.; Ffrench-Constant, C. An integrin-contactin complex regulates CNS myelination by differential Fyn phosphorylation. J. Neurosci. 2009, 29, 9174–9185. [Google Scholar] [CrossRef]
- Gentry, J.J.; Barker, P.A.; Carter, B.D. The p75 neurotrophin receptor: Multiple interactors and numerous functions. Prog. Brain Res. 2004, 146, 25–39. [Google Scholar] [CrossRef]
- Barker, P.A. p75NTR is positively promiscuous: Novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef]
- Lee, X.; Yang, Z.; Shao, Z.; Rosenberg, S.S.; Levesque, M.; Pepinsky, R.B.; Qiu, M.; Miller, R.H.; Chan, J.R.; Mi, S. NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J. Neurosci. 2007, 27, 220–225. [Google Scholar] [CrossRef]
- Domeniconi, M.; Filbin, M.T. Overcoming inhibitors in myelin to promote axonal regeneration. J. Neurol. Sci. 2005, 233, 43–47. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Yamamura, T.; Arima, K.; Konno, H. TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2007, 33, 99–107. [Google Scholar]
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.H.; et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med. 2007, 13, 1228–1233. [Google Scholar] [CrossRef]
- Rudick, R.A.; Mi, S.; Sandrock, A.W., Jr. LINGO-1 antagonists as therapy for multiple sclerosis: In vitro and in vivo evidence. Expert Opin. Biol. Ther. 2008, 8, 1561–1570. [Google Scholar] [CrossRef]
- Chun, S.J.; Rasband, M.N.; Sidman, R.L.; Habib, A.A.; Vartanian, T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J. Cell Biol. 2003, 163, 397–408. [Google Scholar] [CrossRef]
- Kramer, E.M.; Klein, C.; Koch, T.; Boytinck, M.; Trotter, J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J. Biol. Chem. 1999, 274, 29042–29049. [Google Scholar] [CrossRef]
- Relucio, J.; Tzvetanova, I.D.; Ao, W.; Lindquist, S.; Colognato, H. Laminin alters fyn regulatory mechanisms and promotes oligodendrocyte development. J. Neurosci. 2009, 29, 11794–11806. [Google Scholar] [CrossRef]
- Charles, P.; Tait, S.; Faivre-Sarrailh, C.; Barbin, G.; Gunn-Moore, F.; Denisenko-Nehrbass, N.; Guennoc, A.M.; Girault, J.A.; Brophy, P.J.; Lubetzki, C. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr. Biol. 2002, 12, 217–220. [Google Scholar]
- Colognato, H.; Galvin, J.; Wang, Z.; Relucio, J.; Nguyen, T.; Harrison, D.; Yurchenco, P.D.; Ffrench-Constant, C. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development 2007, 134, 1723–1736. [Google Scholar] [CrossRef]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef]
- Wolswijk, G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J. Neurosci. 1998, 18, 601–609. [Google Scholar]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef]
- Zhao, C.; Li, W.W.; Franklin, R.J. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol. Aging 2006, 27, 1298–1307. [Google Scholar] [CrossRef]
- Blakemore, W.F.; Chari, D.M.; Gilson, J.M.; Crang, A.J. Modelling large areas of demyelination in the rat reveals the potential and possible limitations of transplanted glial cells for remyelination in the CNS. Glia 2002, 38, 155–168. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Sloane, J.A. Regulation of remyelination in multiple sclerosis. FEBS Lett. 2011, 585, 3821–3828. [Google Scholar] [CrossRef]
- Talbott, J.F.; Loy, D.N.; Liu, Y.; Qiu, M.S.; Bunge, M.B.; Rao, M.S.; Whittemore, S.R. Endogenous Nkx2.2+/Olig2+ oligodendrocyte precursor cells fail to remyelinate the demyelinated adult rat spinal cord in the absence of astrocytes. Exp. Neurol. 2005, 192, 11–24. [Google Scholar] [CrossRef]
- Miller, R.H.; Mi, S. Dissecting demyelination. Nat. Neurosci. 2007, 10, 1351–1354. [Google Scholar] [CrossRef]
- Franklin, R.J.; Kotter, M.R. The biology of CNS remyelination: The key to therapeutic advances. J. Neurol. 2008, 255, 19–25. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Hoglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef]
- Nakahara, T.; Urabe, K.; Fukagawa, S.; Uchi, H.; Inaba, K.; Furue, M.; Moroi, Y. Engagement of human monocyte-derived dendritic cells into interleukin (IL)-12 producers by IL-1β + interferon (IFN)-gamma. Clin. Exp. Immunol. 2005, 139, 476–482. [Google Scholar] [CrossRef]
- Petermann, F.; Korn, T. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett. 2011, 585, 3747–3757. [Google Scholar] [CrossRef]
- Kroenke, M.A.; Carlson, T.J.; Andjelkovic, A.V.; Segal, B.M. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J. Exp. Med. 2008, 205, 1535–1541. [Google Scholar] [CrossRef]
- Kang, Z.; Altuntas, C.Z.; Gulen, M.F.; Liu, C.; Giltiay, N.; Qin, H.; Liu, L.; Qian, W.; Ransohoff, R.M.; Bergmann, C.; et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 2010, 32, 414–425. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Yu, C.; Argyropoulos, G.; Zhang, Y.; Kastin, A.J.; Hsuchou, H.; Pan, W. Neuroinflammation activates Mdr1b efflux transport through NFkappaB: Promoter analysis in BBB endothelia. Cell Physiol. Biochem. 2008, 22, 745–756. [Google Scholar] [CrossRef]
- Vela, J.M.; Molina-Holgado, E.; Arevalo-Martin, A.; Almazan, G.; Guaza, C. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol. Cell. Neurosci. 2002, 20, 489–502. [Google Scholar] [CrossRef]
- Meeuwsen, S.; Persoon-Deen, C.; Bsibsi, M.; Ravid, R.; van Noort, J.M. Cytokine, chemokine and growth factor gene profiling of cultured human astrocytes after exposure to proinflammatory stimuli. Glia 2003, 43, 243–253. [Google Scholar] [CrossRef]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef]
- Lassmann, H.; Ransohoff, R.M. The CD4-Th1 model for multiple sclerosis: A critical (correction of crucial) re-appraisal. Trends Immunol. 2004, 25, 132–137. [Google Scholar] [CrossRef]
- Issazadeh, S.; Mustafa, M.; Ljungdahl, A.; Hojeberg, B.; Dagerlind, A.; Elde, R.; Olsson, T. Interferon gamma, interleukin 4 and transforming growth factor beta in experimental autoimmune encephalomyelitis in Lewis rats: Dynamics of cellular mRNA expression in the central nervous system and lymphoid cells. J. Neurosci. Res. 1995, 40, 579–590. [Google Scholar] [CrossRef]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6–gp130–STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Matusevicius, D.; Kivisakk, P.; He, B.; Kostulas, N.; Ozenci, V.; Fredrikson, S.; Link, H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult. Scler. 1999, 5, 101–104. [Google Scholar]
- Craner, M.J.; Hains, B.C.; Lo, A.C.; Black, J.A.; Waxman, S.G. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain 2004, 127, 294–303. [Google Scholar] [CrossRef]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar]
- Wu, H.Y.; Tomizawa, K.; Matsui, H. Calpain-calcineurin signaling in the pathogenesis of calcium-dependent disorder. Acta Med. Okayama 2007, 61, 123–137. [Google Scholar]
- Banik, N.L.; Chakrabarti, A.K.; Konat, G.W.; Gantt-Wilford, G.; Hogan, E.L. Calcium-activated neutral proteinase (calpain) activity in C6 cell line: Compartmentation of mu and m calpain. J. Neurosci. Res. 1992, 31, 708–714. [Google Scholar] [CrossRef]
- Chakrabarti, A.K.; Dasgupta, S.; Banik, N.L.; Hogan, E.L. Ganglioside-modulated proteolysis by Ca2+-activated neutral proteinase (CANP): A role of glycoconjugates in CANP regulation. J. Neurochem. 1990, 54, 1816–1819. [Google Scholar] [CrossRef]
- Chakrabarti, A.K.; Dasgupta, S.; Banik, N.L.; Hogan, E.L. Regulation of the calcium-activated neutral proteinase (CANP) of bovine brain by myelin lipids. Biochim. Biophys. Acta 1990, 1038, 195–198. [Google Scholar]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Siriwardena, D.; Serraino, I.; Dugo, L.; Britti, D.; Mazzullo, G.; Caputi, A.P.; Thiemermann, C. Calpain inhibitor I reduces the development of acute and chronic inflammation. Am. J. Pathol. 2000, 157, 2065–2079. [Google Scholar] [CrossRef]
- Butler, J.T.; Samantaray, S.; Beeson, C.C.; Ray, S.K.; Banik, N.L. Involvement of calpain in the process of Jurkat T cell chemotaxis. J. Neurosci. Res. 2009, 87, 626–635. [Google Scholar] [CrossRef]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Pant, H.C. Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell. Mol. Neurobiol. 2009, 29, 1073–1080. [Google Scholar] [CrossRef]
- Gill, M.B.; Perez-Polo, J.R. Bax shuttling after rotenone treatment of neuronal primary cultures: Effects on cell death phenotypes. J. Neurosci. Res. 2009, 87, 2047–2065. [Google Scholar] [CrossRef]
- Guyton, M.K.; Wingrave, J.M.; Yallapragada, A.V.; Wilford, G.G.; Sribnick, E.A.; Matzelle, D.D.; Tyor, W.R.; Ray, S.K.; Banik, N.L. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J. Neurosci. Res. 2005, 81, 53–61. [Google Scholar] [CrossRef]
- Shields, D.C.; Avgeropoulos, N.G.; Banik, N.L.; Tyor, W.R. Acute multiple sclerosis characterized by extensive mononuclear phagocyte infiltration. Neurochem. Res. 2000, 25, 1517–1520. [Google Scholar] [CrossRef]
- Hassen, G.W.; Feliberti, J.; Kesner, L.; Stracher, A.; Mokhtarian, F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 180, 135–146. [Google Scholar] [CrossRef]
- Bennett, J.L.; Stuve, O. Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: Therapeutic implications. Clin. Neuropharmacol. 2009, 32, 121–132. [Google Scholar] [CrossRef]
- Imam, S.A.; Guyton, M.K.; Haque, A.; Vandenbark, A.; Tyor, W.R.; Ray, S.K.; Banik, N.L. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J. Neuroimmunol. 2007, 190, 139–145. [Google Scholar] [CrossRef]
- Smith, A.W.; Doonan, B.P.; Tyor, W.R.; Abou-Fayssal, N.; Haque, A.; Banik, N.L. Regulation of Th1/Th17 cytokines and IDO gene expression by inhibition of calpain in PBMCs from MS patients. J. Neuroimmunol. 2011, 232, 179–185. [Google Scholar] [CrossRef]
- Shields, D.C.; Banik, N.L. Pathophysiological role of calpain in experimental demyelination. J. Neurosci. Res. 1999, 55, 533–541. [Google Scholar] [CrossRef]
- Hassen, G.W.; Feliberti, J.; Kesner, L.; Stracher, A.; Mokhtarian, F. Prevention of axonal injury using calpain inhibitor in chronic progressive experimental autoimmune encephalomyelitis. Brain Res. 2008, 1236, 206–215. [Google Scholar] [CrossRef]
- Das, A.; Guyton, M.K.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of lewis rats during development of acute experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2008, 86, 2992–3001. [Google Scholar] [CrossRef]
- Guyton, M.K.; Das, A.; Samantaray, S.; Wallace, G.C.; Butler, J.T.; Ray, S.K.; Banik, N.L. Calpeptin Attenuated Inflammation, Cell Death, and Axonal Damage in Animal Model of Multiple Sclerosis. J. Neurosci. Res. 2010, 88, 2398–2408. [Google Scholar]
- Guyton, M.K.; Brahmachari, S.; Das, A.; Samantaray, S.; Inoue, J.; Azuma, M.; Ray, S.K.; Banik, N.L. Inhibition of calpain attenuates encephalitogenicity of MBP-specific T cells. J. Neurochem. 2009, 110, 1895–1907. [Google Scholar] [CrossRef]
- Adorini, L. Antigen-based immunointervention in human autoimmune diseases. Trends Biotechnol. 2004, 22, 547–549. [Google Scholar] [CrossRef]
- Jahng, A.W.; Maricic, I.; Pedersen, B.; Burdin, N.; Naidenko, O.; Kronenberg, M.; Koezuka, Y.; Kumar, V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 194, 1789–1799. [Google Scholar] [CrossRef]
- O'Keeffe, J.; Gately, C.M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L. T-cells expressing natural killer (NK) receptors are altered in multiple sclerosis and responses to alpha-galactosylceramide are impaired. J. Neurol. Sci. 2008, 275, 22–28. [Google Scholar] [CrossRef]
- Gately, C.M.; Podbielska, M.; Counihan, T.; Hennessy, M.; Leahy, T.; Moran, A.P.; Hogan, E.L.; O’Keeffe, J. Invariant Natural Killer T-cell anergy to endogenous myelin acetyl-glycolipids in multiple sclerosis. J. Neuroimmunol. 2013, 259, 1–7. [Google Scholar] [CrossRef]
- Oldstone, M.B. Molecular mimicry, microbial infection, and autoimmune disease: Evolution of the concept. Curr. Top. Microbiol. Immunol. 2005, 296, 1–17. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Allen, P.M.; Celada, F.; Cohen, I.R.; De Boer, R.; Garcia, K.C.; Goldstein, B.; Greenspan, R.; Hafler, D.; Hodgkin, P.; et al. Polyspecificity of T cell and B cell receptor recognition. Semin. Immunol. 2007, 19, 216–224. [Google Scholar] [CrossRef]
- Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci. 2009, 278, 5–15. [Google Scholar] [CrossRef]
- Podbielska, M.; Krotkiewski, H.; Hogan, E.L. Signaling and regulatory functions of bioactive sphingolipids as therapeutic targets in multiple sclerosis. Neurochem. Res. 2012, 37, 1154–1169. [Google Scholar] [CrossRef]
- Taniguchi, M.; Tashiro, T.; Dashtsoodol, N.; Hongo, N.; Watarai, H. The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int. Immunol. 2010, 22, 1–6. [Google Scholar] [CrossRef]
- Porcelli, S.; Yockey, C.E.; Brenner, M.B.; Balk, S.P. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8-α/β T cells demonstrates preferential use of several V β genes and an invariant TCR alpha chain. J. Exp. Med. 1993, 178, 1–16. [Google Scholar] [CrossRef]
- Borg, N.; Holland, M. The effect of glycosaminoglycans on rat gametes in vitro and the associated signal pathway. Reproduction 2008, 135, 311–319. [Google Scholar] [CrossRef]
- Scott-Browne, J.P.; Matsuda, J.L.; Mallevaey, T.; White, J.; Borg, N.A.; McCluskey, J.; Rossjohn, J.; Kappler, J.; Marrack, P.; Gapin, L. Germline-encoded recognition of diverse glycolipids by natural killer T cells. Nat. Immunol. 2007, 8, 1105–1113. [Google Scholar] [CrossRef]
- Hogan, E.L.; Podbielska, M.; O’Keeffe, J. Implications of lymphocyte anergy to glycolipids in multiple sclerosis (MS): iNKT cells may mediate the MS infectious trigger. J. Clin. Cell. Immunol. 2013, 4, 144. [Google Scholar] [CrossRef]
- Cui, J.; Shin, T.; Kawano, T.; Sato, H.; Kondo, E.; Toura, I.; Kaneko, Y.; Koseki, H.; Kanno, M.; Taniguchi, M. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 1997, 278, 1623–1626. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Rossjohn, J. New ways to turn on NKT cells. J. Exp. Med. 2011, 208, 1121–1125. [Google Scholar] [CrossRef]
- Ito, K.; Karasawa, M.; Kawano, T.; Akasaka, T.; Koseki, H.; Akutsu, Y.; Kondo, E.; Sekiya, S.; Sekikawa, K.; Harada, M.; et al. Involvement of decidual Valpha14 NKT cells in abortion. Proc. Natl. Acad. Sci. USA 2000, 97, 740–744. [Google Scholar] [CrossRef]
- Foote, A.K.; Blakemore, W.F. Inflammation stimulates remyelination in areas of chronic demyelination. Brain 2005, 128, 528–539. [Google Scholar] [CrossRef]
- Chari, D.M.; Blakemore, W.F. New insights into remyelination failure in multiple sclerosis: Implications for glial cell transplantation. Mult. Scler. 2002, 8, 271–277. [Google Scholar] [CrossRef]
- Ben-Hur, T.; Goldman, S.A. Prospects of cell therapy for disorders of myelin. Ann. N. Y. Acad. Sci. 2008, 1142, 218–249. [Google Scholar] [CrossRef]
- Warrington, A.E.; Bieber, A.J.; Ciric, B.; Van Keulen, V.; Pease, L.R.; Mitsunaga, Y.; Paz Soldan, M.M.; Rodriguez, M. Immunoglobulin-mediated CNS repair. J. Allergy Clin. Immunol. 2001, 108, S121–S125. [Google Scholar] [CrossRef]
- Warrington, A.E.; Rodriguez, M. Remyelination-promoting human IgMs: Developing a therapeutic reagent for demyelinating disease. Curr. Top. Microbiol. Immunol. 2008, 318, 213–239. [Google Scholar] [CrossRef]
- Paz Soldan, M.M.; Warrington, A.E.; Bieber, A.J.; Ciric, B.; Van Keulen, V.; Pease, L.R.; Rodriguez, M. Remyelination-promoting antibodies activate distinct Ca2+ influx pathways in astrocytes and oligodendrocytes: Relationship to the mechanism of myelin repair. Mol. Cell. Neurosci. 2003, 22, 14–24. [Google Scholar] [CrossRef]
- Lang, W.; Rodriguez, M.; Lennon, V.A.; Lampert, P.W. Demyelination and remyelination in murine viral encephalomyelitis. Ann. N. Y. Acad. Sci. 1984, 436, 98–102. [Google Scholar]
- Huang, D.W.; McKerracher, L.; Braun, P.E.; David, S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron 1999, 24, 639–647. [Google Scholar] [CrossRef]
- Rodriguez, M.; Lennon, V.A.; Benveniste, E.N.; Merrill, J.E. Remyelination by oligodendrocytes stimulated by antiserum to spinal cord. J. Neuropathol. Exp. Neurol. 1987, 46, 84–95. [Google Scholar] [CrossRef]
- Warrington, A.E.; Asakura, K.; Bieber, A.J.; Ciric, B.; Van Keulen, V.; Kaveri, S.V.; Kyle, R.A.; Pease, L.R.; Rodriguez, M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 6820–6825. [Google Scholar] [CrossRef]
- Bieber, A.J.; Warrington, A.; Asakura, K.; Ciric, B.; Kaveri, S.V.; Pease, L.R.; Rodriguez, M. Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia 2002, 37, 241–249. [Google Scholar] [CrossRef]
- Pavelko, K.D.; van Engelen, B.G.; Rodriguez, M. Acceleration in the rate of CNS remyelination in lysolecithin-induced demyelination. J. Neurosci. 1998, 18, 2498–2505. [Google Scholar]
- Miller, D.J.; Bright, J.J.; Sriram, S.; Rodriguez, M. Successful treatment of established relapsing experimental autoimmune encephalomyelitis in mice with a monoclonal natural autoantibody. J. Neuroimmunol. 1997, 75, 204–209. [Google Scholar] [CrossRef]
- Miller, D.J.; Sanborn, K.S.; Katzmann, J.A.; Rodriguez, M. Monoclonal autoantibodies promote central nervous system repair in an animal model of multiple sclerosis. J. Neurosci. 1994, 14, 6230–6238. [Google Scholar]
- Asakura, K.; Miller, D.J.; Murray, K.; Bansal, R.; Pfeiffer, S.E.; Rodriguez, M. Monoclonal autoantibody SCH94.03, which promotes central nervous system remyelination, recognizes an antigen on the surface of oligodendrocytes. J. Neurosci. Res. 1996, 43, 273–281. [Google Scholar] [CrossRef]
- Mitsunaga, Y.; Ciric, B.; Van Keulen, V.; Warrington, A.E.; Paz Soldan, M.; Bieber, A.J.; Rodriguez, M.; Pease, L.R. Direct evidence that a human antibody derived from patient serum can promote myelin repair in a mouse model of chronic-progressive demyelinating disease. FASEB J. 2002, 16, 1325–1327. [Google Scholar]
- Rodriguez, M.; Warrington, A.E.; Pease, L.R. Invited Article: Human natural autoantibodies in the treatment of neurologic disease. Neurology 2009, 72, 1269–1276. [Google Scholar] [CrossRef]
- Duncan, I.D. Glial cell transplantation and remyelination of the central nervous system. Neuropathol. Appl. Neurobiol. 1996, 22, 87–100. [Google Scholar] [CrossRef]
- Howe, C.L.; Bieber, A.J.; Warrington, A.E.; Pease, L.R.; Rodriguez, M. Antiapoptotic signaling by a remyelination-promoting human antimyelin antibody. Neurobiol. Dis. 2004, 15, 120–131. [Google Scholar] [CrossRef]
- Warrington, A.E.; Bieber, A.J.; Ciric, B.; Pease, L.R.; Van Keulen, V.; Rodriguez, M. A recombinant human IgM promotes myelin repair after a single, very low dose. J. Neurosci. Res. 2007, 85, 967–976. [Google Scholar] [CrossRef]
- Pirko, I.; Ciric, B.; Gamez, J.; Bieber, A.J.; Warrington, A.E.; Johnson, A.J.; Hanson, D.P.; Pease, L.R.; Macura, S.I.; Rodriguez, M. A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. FASEB J. 2004, 18, 1577–1579. [Google Scholar]
- Zemann, B.; Kinzel, B.; Muller, M.; Reuschel, R.; Mechtcheriakova, D.; Urtz, N.; Bornancin, F.; Baumruker, T.; Billich, A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 2006, 107, 1454–1458. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Hartmann, S.; Bartlett, M.; Riviere, G.J.; Neddermann, D.; Wang, Y.; Port, A.; Schmouder, R.L. Oral-intravenous crossover study of fingolimod pharmacokinetics, lymphocyte responses and cardiac effects. Biopharm. Drug Dispos. 2007, 28, 97–104. [Google Scholar] [CrossRef]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef]
- Kahan, B.D.; Karlix, J.L.; Ferguson, R.M.; Leichtman, A.B.; Mulgaonkar, S.; Gonwa, T.A.; Skerjanec, A.; Schmouder, R.L.; Chodoff, L. Pharmacodynamics, pharmacokinetics, and safety of multiple doses of FTY720 in stable renal transplant patients: A multicenter, randomized, placebo-controlled, phase I study. Transplantation 2003, 76, 1079–1084. [Google Scholar] [CrossRef]
- Budde, K.; Schmouder, R.L.; Nashan, B.; Brunkhorst, R.; Lücker, P.W.; Mayer, T.; Brookman, L.; Nedelman, J.; Skerjanec, A.; Bohler, T.; Neumayer, H.H. Pharmacodynamics of single doses of the novel immunosuppressant FTY720 in stable renal transplant patients. Am. J. Transplant. 2003, 3, 846–854. [Google Scholar] [CrossRef]
- Lee, C.W.; Choi, J.W.; Chun, J. Neurological S1P signaling as an emerging mechanism of action of oral FTY720 (fingolimod) in multiple sclerosis. Arch. Pharm. Res. 2010, 33, 1567–1574. [Google Scholar] [CrossRef]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar]
- Foster, C.A.; Howard, L.M.; Schweitzer, A.; Persohn, E.; Hiestand, P.C.; Balatoni, B.; Reuschel, R.; Beerli, C.; Schwartz, M.; Billich, A. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: Consequences for mode of action in multiple sclerosis. J. Pharmacol. Exp. Ther. 2007, 323, 469–475. [Google Scholar] [CrossRef]
- Fujino, M.; Funeshima, N.; Kitazawa, Y.; Kimura, H.; Amemiya, H.; Suzuki, S.; Li, X.K. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J. Pharmacol. Exp. Ther. 2003, 305, 70–77. [Google Scholar] [CrossRef]
- Webb, M.; Tham, C.S.; Lin, F.F.; Lariosa-Willingham, K.; Yu, N.; Hale, J.; Mandala, S.; Chun, J.; Rao, T.S. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J. Neuroimmunol. 2004, 153, 108–121. [Google Scholar] [CrossRef]
- Kataoka, H.; Sugahara, K.; Shimano, K.; Teshima, K.; Koyama, M.; Fukunari, A.; Chiba, K. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell. Mol. Immunol. 2005, 2, 439–448. [Google Scholar]
- Balatoni, B.; Storch, M.K.; Swoboda, E.M.; Schonborn, V.; Koziel, A.; Lambrou, G.N.; Hiestand, P.C.; Weissert, R.; Foster, C.A. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res. Bull. 2007, 74, 307–316. [Google Scholar] [CrossRef]
- Miron, V.E.; Hall, J.A.; Kennedy, T.E.; Soliven, B.; Antel, J.P. Cyclical and dose-dependent responses of adult human mature oligodendrocytes to fingolimod. Am. J. Pathol. 2008, 173, 1143–1152. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.W.; O'Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Gold, R.; Giovannoni, G.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; Robinson, R.; Riester, K.; Rana, J.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): A randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 2167–2175. [Google Scholar] [CrossRef]
- Klotz, L.; Wiendl, H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin. Biol. Ther. 2013, 13, 831–846. [Google Scholar] [CrossRef]
- Ali, R.; Nicholas, R.S.; Muraro, P.A. Drugs in development for relapsing multiple sclerosis. Drugs 2013, 73, 625–650. [Google Scholar] [CrossRef]
- Huang, J.K.; Franklin, R.J. Current status of myelin replacement therapies in multiple sclerosis. Prog. Brain Res. 2012, 201, 219–231. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Podbielska, M.; Banik, N.L.; Kurowska, E.; Hogan, E.L. Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination. Brain Sci. 2013, 3, 1282-1324. https://doi.org/10.3390/brainsci3031282
Podbielska M, Banik NL, Kurowska E, Hogan EL. Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination. Brain Sciences. 2013; 3(3):1282-1324. https://doi.org/10.3390/brainsci3031282
Chicago/Turabian StylePodbielska, Maria, Naren L. Banik, Ewa Kurowska, and Edward L. Hogan. 2013. "Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination" Brain Sciences 3, no. 3: 1282-1324. https://doi.org/10.3390/brainsci3031282
APA StylePodbielska, M., Banik, N. L., Kurowska, E., & Hogan, E. L. (2013). Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination. Brain Sciences, 3(3), 1282-1324. https://doi.org/10.3390/brainsci3031282