The Role of Ghrelin in Neuroprotection after Ischemic Brain Injury

Abstract
:1. Introduction
2. Ghrelin’s Role in Neuroprotection
2.1. Parkinson’s and Alzheimer’s Diseases
2.2. Cerebral Ischemia
2.2.1. Observations of Neuroprotection
2.2.2. Mechanisms of Neuroprotection
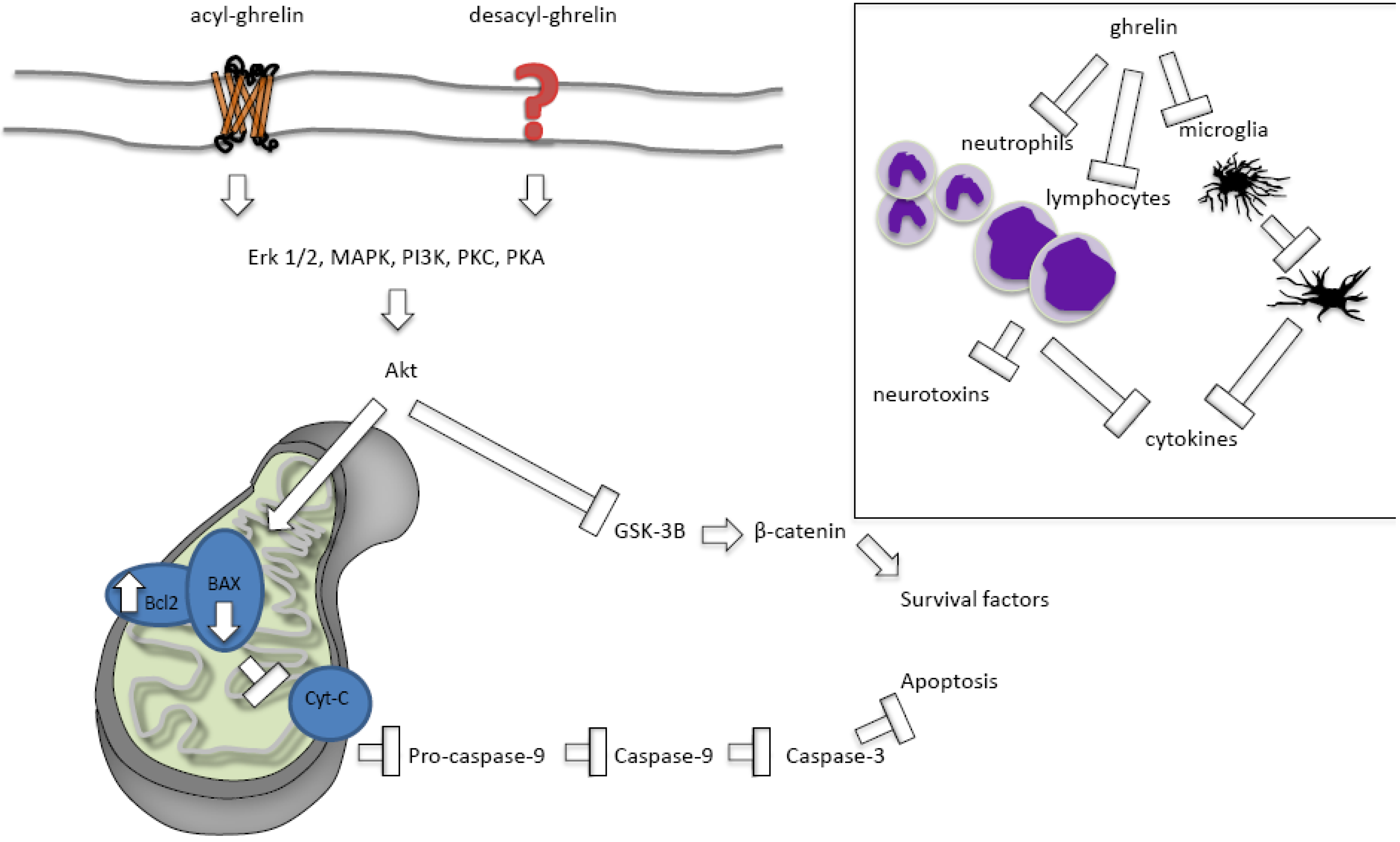
2.2.2.1. Ghrelin Protects against Apoptosis
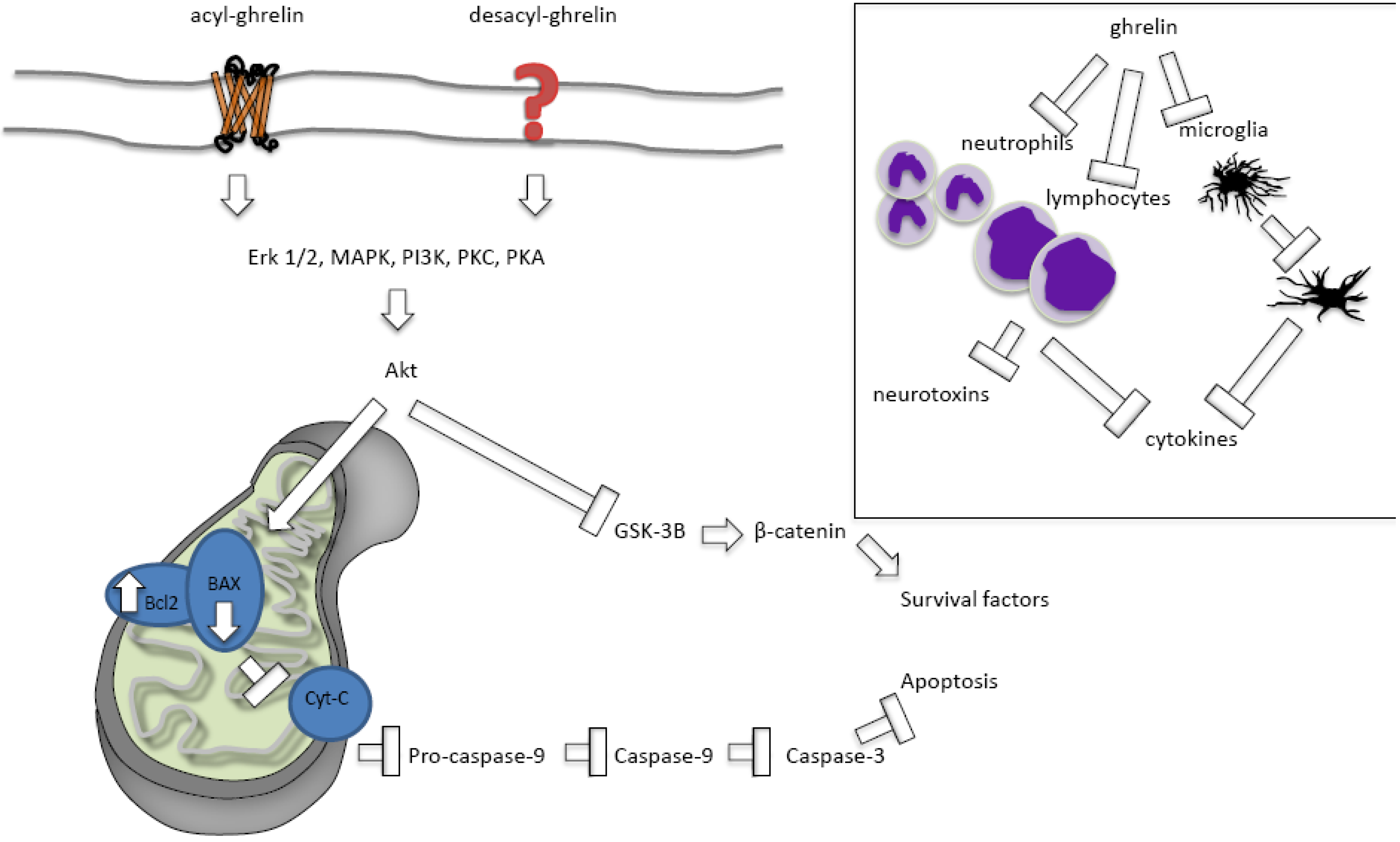
2.2.2.2. Ghrelin’s Effects on Necrosis and Autophagy
2.2.2.3. Acylated versus Des-Acylated Ghrelin
2.2.2.4. Ghrelin Protects against Inflammation
3. Clinical Considerations

{kind=link}
| Ischemic injury | Dose | Timing | Effect | Mechanism of action | Reference |
|---|---|---|---|---|---|
| Oxygen-glucose deprivation in cultured cells | Acylated and unacylated ghrelin, 100 nM each | Pretreatment for 24 h | ↓ cell death (both) | ↓ apoptosis | [56,62] |
| Doxorubicin H9c2 cardiomyocytes | Total ghrelin, 1 µM | Co-treatment for 24 h | ↓ cell death (both) | ↓ apoptosis GHSR-1a-independent mechanism | [31] |
| 4VO forebrain ischemia/reperfusion | Total ghrelin, i.p., 0.4 mg/kg | Daily for 3 days post injury | ↑ cell survival CA1 hippocampus | ↓ apoptosis | [44] |
| MCAO Focal ischemia/reperfusion | Total ghrelin, i.v., 10 pmol/kg | Immediately post injury | ↓cortical neuron injury | ↓ apoptosis ↑ expression of GHSR-1a | [45] |
| MCAO | Total ghrelin, i.v., ~7 pmol/kg | Immediately post injury infusion for 1 h | ↓ neurological deficit, ↓ infarct size at 24 h and 7 days | ↓ apoptosis ↓ inflammation | [46] |
| MCAO | Total (80 µg/kg) or desacyl (160 µg/kg), i.p. | 30 min prior to injury and immediately post | ↓ cortical neuron injury (both) | ↓ apoptosis GHSR-1a-independent mechanism | [59] |
| Neonatal hypoxia-ischemia | GHS-hexarelin, icv, 1 µg in 5 µL | Immediately post injury | ↓ cortical, hippocampal, thalamic injury, ↔ striatum | ↓ apoptosis | [43] |
| Spinal cord ischemia/reperfusion | Total ghrelin, i.p., 100 µg/kg | Ischemia onset | ↑ neurological scores | ↓ apoptosis ↓ inflammation ↑ expression of GHSR-1a | [65] |
| Subarachnoid hemorrhage | Total ghrelin, i.p., 10 µg/kg/day | Immediately post injury and 24 h later | ↑ neurological scores | ↓ inflammation | [74] |
| Traumatic brain injury | Total ghrelin, i.v., 4, 8 or 16 nmol/rat | 45 min post-injury | ↓ cortical neuron injury ↓ behavioural deficits | ↓ apoptosis ↓ inflammation | [73] |
| Traumatic brain injury | Total ghrelin, i.p., 10 µg/kg/dose | Immediately prior to and 1 h post injury | ↓ cell death | ↓ inflammation ↓ blood brain barrier permeability | [58] |
4. Conclusions
References
- Papadopoulos, S.M.; Chandler, W.F.; Salamat, M.S.; Topol, E.J.; Sackellares, J.C. Recombinant human tissue-type plasminogen activator therapy in acute thromboembolic stroke. J. Neurosurg. 1987, 67, 394–398. [Google Scholar] [CrossRef]
- Demers, G.; Meurer, W.J.; Shih, R.; Rosenbaum, S.; Vilke, G.M. Tissue Plasminogen Activator and Stroke: Review of the Literature for the Clinician. J. Emerg. Med. 2012, 43, 1149–1154. [Google Scholar] [CrossRef]
- World Health Organizition (WHO). The top 10 causes of death. Available online: http://www.who.int/mediacentre/factsheets/fs310/en/index.html (accessed on 9 December 2012).
- Lago, F.; Gonzalez-Juanatey, J.R.; Casanueva, F.F.; Gomez-Reino, J.; Dieguez, C.; Gualillo, O. Ghrelin, the same peptide for different functions: Player or bystander? Vitam. Horm. 2005, 71, 405–432. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Solenberg, P.J.; Perkins, D.R.; Willency, J.A.; Knierman, M.D.; Jin, Z.; Witcher, D.R.; Luo, S.; Onyia, J.E.; Hale, J.E. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl. Acad. Sci. USA 2008, 105, 6320–6325. [Google Scholar] [CrossRef]
- Cummings, D.E. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol. Behav. 2006, 89, 71–84. [Google Scholar] [CrossRef]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschop, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Briggs, D.I.; Andrews, Z.B. Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 2011, 93, 48–57. [Google Scholar] [CrossRef]
- Andrews, Z.B. Central mechanisms involved in the orexigenic actions of ghrelin. Peptides 2011, 32, 2248–2255. [Google Scholar]
- Abizaid, A.; Liu, Z.W.; Andrews, Z.B.; Shanabrough, M.; Borok, E.; Elsworth, J.D.; Roth, R.H.; Sleeman, M.W.; Picciotto, M.R.; Tschop, M.H.; et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 2006, 116, 3229–3239. [Google Scholar] [CrossRef]
- Naleid, A.M.; Grace, M.K.; Cummings, D.E.; Levine, A.S. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides 2005, 26, 2274–2279. [Google Scholar] [CrossRef]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A.; et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef]
- Spencer, S.J.; Xu, L.; Clarke, M.A.; Lemus, M.; Reichenbach, A.; Geenen, B.; Kozicz, T.; Andrews, Z.B. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol. Psychiatry 2012, 72, 457–465. [Google Scholar] [CrossRef]
- Lutter, M.; Sakata, I.; Osborne-Lawrence, S.; Rovinsky, S.A.; Anderson, J.G.; Jung, S.; Birnbaum, S.; Yanagisawa, M.; Elmquist, J.K.; Nestler, E.J.; Zigman, J.M. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 2008, 11, 752–753. [Google Scholar] [CrossRef]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef]
- Guan, X.M.; Yu, H.; Palyha, O.C.; McKee, K.K.; Feighner, S.D.; Sirinathsinghji, D.J.; Smith, R.G.; van der Ploeg, L.H.; Howard, A.D. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 1997, 48, 23–29. [Google Scholar] [CrossRef]
- Bennett, W.L.; Keeton, A.B.; Ji, S.; Xu, J.; Messina, J.L. Insulin regulation of growth hormone receptor gene expression: Involvement of both the PI-3 kinase and MEK/ERK signaling pathways. Endocrine 2007, 32, 219–226. [Google Scholar] [CrossRef]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar]
- Camina, J.P. Cell biology of the ghrelin receptor. J. Neuroendocrinol. 2006, 18, 65–76. [Google Scholar] [CrossRef]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Liberator, P.A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, D.L.; Palyha, O.C.; Anderson, J.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar]
- McKee, K.K.; Palyha, O.C.; Feighner, S.D.; Hreniuk, D.L.; Tan, C.P.; Phillips, M.S.; Smith, R.G.; van der Ploeg, L.H.; Howard, A.D. Molecular analysis of rat pituitary and hypothalamic growth hormone secretagogue receptors. Mol. Endocrinol. 1997, 11, 415–423. [Google Scholar] [CrossRef]
- Petersen, P.S.; Woldbye, D.P.; Madsen, A.N.; Egerod, K.L.; Jin, C.; Lang, M.; Rasmussen, M.; Beck-Sickinger, A.G.; Holst, B. In vivo characterization of high Basal signaling from the ghrelin receptor. Endocrinology 2009, 150, 4920–4930. [Google Scholar] [CrossRef]
- Kern, A.; Albarran-Zeckler, R.; Walsh, H.E.; Smith, R.G. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 2012, 73, 317–332. [Google Scholar] [CrossRef]
- Rediger, A.; Piechowski, C.L.; Yi, C.X.; Tarnow, P.; Strotmann, R.; Gruters, A.; Krude, H.; Schoneberg, T.; Tschop, M.H.; Kleinau, G.; Biebermann, H. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J. Biol. Chem. 2011, 286, 39623–39631. [Google Scholar] [CrossRef]
- Banks, W.A.; Tschop, M.; Robinson, S.M.; Heiman, M.L. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 2002, 302, 822–827. [Google Scholar] [CrossRef]
- Muccioli, G.; Tschop, M.; Papotti, M.; Deghenghi, R.; Heiman, M.; Ghigo, E. Neuroendocrine and peripheral activities of ghrelin: Implications in metabolism and obesity. Eur. J. Pharmacol. 2002, 440, 235–254. [Google Scholar] [CrossRef]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef]
- Chang, L.; Ren, Y.; Liu, X.; Li, W.G.; Yang, J.; Geng, B.; Weintraub, N.L.; Tang, C. Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 2004, 43, 165–170. [Google Scholar] [CrossRef]
- Frascarelli, S.; Ghelardoni, S.; Ronca-Testoni, S.; Zucchi, R. Effect of ghrelin and synthetic growth hormone secretagogues in normal and ischemic rat heart. Basic Res. Cardiol. 2003, 98, 401–405. [Google Scholar] [CrossRef]
- Andrews, Z.B. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci. 2010, 34, 31–40. [Google Scholar] [CrossRef]
- Jiang, H.; Li, L.J.; Wang, J.; Xie, J.X. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 2008, 212, 532–537. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschoep, M.; et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef]
- Moon, M.; Kim, H.G.; Hwang, L.; Seo, J.H.; Kim, S.; Hwang, S.; Lee, D.; Chung, H.; Oh, M.S.; Lee, K.T.; Park, S. Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease by blocking microglial activation. Neurotox. Res. 2009, 15, 332–347. [Google Scholar] [CrossRef]
- Unger, M.M.; Moller, J.C.; Mankel, K.; Eggert, K.M.; Bohne, K.; Bodden, M.; Stiasny-Kolster, K.; Kann, P.H.; Mayer, G.; Tebbe, J.J.; Oertel, W.H. Postprandial ghrelin response is reduced in patients with Parkinson’s disease and idiopathic REM sleep behaviour disorder: A peripheral biomarker for early Parkinson’s disease? J. Neurol. 2011, 258, 982–990. [Google Scholar] [CrossRef]
- Proto, C.; Romualdi, D.; Cento, R.M.; Spada, R.S.; Di Mento, G.; Ferri, R.; Lanzone, A. Plasma levels of neuropeptides in Alzheimer’s disease. Gynecol. Endocrinol. 2006, 22, 213–218. [Google Scholar] [CrossRef]
- Gahete, M.D.; Rubio, A.; Cordoba-Chacon, J.; Gracia-Navarro, F.; Kineman, R.D.; Avila, J.; Luque, R.M.; Castano, J.P. Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 2010, 22, 819–828. [Google Scholar]
- Moon, M.; Choi, J.G.; Nam, D.W.; Hong, H.S.; Choi, Y.J.; Oh, M.S.; Mook-Jung, I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1-42 oligomer-injected mice. J. Alzheimers Dis. 2011, 23, 147–159. [Google Scholar]
- Chen, Y.; Cao, C.P.; Li, C.R.; Wang, W.; Zhang, D.; Han, L.L.; Zhang, X.Q.; Kim, A.; Kim, S.; Liu, G.L. Ghrelin modulates insulin sensitivity and tau phosphorylation in high glucose-induced hippocampal neurons. Biol. Pharm. Bull. 2010, 33, 1165–1169. [Google Scholar] [CrossRef]
- Brywe, K.G.; Leverin, A.L.; Gustavsson, M.; Mallard, C.; Granata, R.; Destefanis, S.; Volante, M.; Hagberg, H.; Ghigo, E.; Isgaard, J. Growth hormone-releasing peptide hexarelin reduces neonatal brain injury and alters Akt/glycogen synthase kinase-3beta phosphorylation. Endocrinology 2005, 146, 4665–4672. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, P.S.; Xie, D.; Liu, K.; Chen, L. Ghrelin reduces injury of hippocampal neurons in a rat model of cerebral ischemia/reperfusion. Chin. J. Physiol. 2006, 49, 244–250. [Google Scholar]
- Miao, Y.; Xia, Q.; Hou, Z.; Zheng, Y.; Pan, H.; Zhu, S. Ghrelin protects cortical neuron against focal ischemia/reperfusion in rats. Biochem. Biophys. Res. Commun. 2007, 359, 795–800. [Google Scholar] [CrossRef]
- Cheyuo, C.; Wu, R.; Zhou, M.; Jacob, A.; Coppa, G.; Wang, P. Ghrelin suppresses inflammation and neuronal nitric oxide synthase in focal cerebral ischemia via the vagus nerve. Shock 2011, 35, 258–265. [Google Scholar] [CrossRef]
- Kantorova, E.; Chomova, M.; Kurca, E.; Sivak, S.; Zelenak, K.; Kucera, P.; Galajda, P. Leptin, adiponectin and ghrelin, new potential mediators of ischemic stroke. Neuro Endocrinol. Lett. 2011, 32, 716–721. [Google Scholar]
- Xu, M.; Zhang, H.L. Death and survival of neuronal and astrocytic cells in ischemic brain injury: A role of autophagy. Acta Pharmacol. Sin. 2011, 32, 1089–1099. [Google Scholar] [CrossRef]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef]
- Colbourne, F.; Auer, R.N. Transient Global Cerebral Ischemia Produces Morphologically Necrotic, Not Apoptotic Neurons. In Acute Neuronal Injury: The Role of Excitotoxic Programmed Cell Death Mechanisms; Fujikawa, D.G., Ed.; Springer Science + Business Media: Berlin, Germany, 2010. [Google Scholar]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, Y.; Krieglstein, J.; Mattson, M.P. Evidence for the involvement of Par-4 in ischemic neuron cell death. J. Cereb. Blood Flow Metab. 2001, 21, 334–343. [Google Scholar]
- Shi, Y. A structural view of mitochondria-mediated apoptosis. Nat. Struct. Biol. 2001, 8, 394–401. [Google Scholar] [CrossRef]
- Yoshida, H.; Kong, Y.Y.; Yoshida, R.; Elia, A.J.; Hakem, A.; Hakem, R.; Penninger, J.M.; Mak, T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94, 739–750. [Google Scholar] [CrossRef]
- Offen, D.; Beart, P.M.; Cheung, N.S.; Pascoe, C.J.; Hochman, A.; Gorodin, S.; Melamed, E.; Bernard, R.; Bernard, O. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 5789–5794. [Google Scholar] [CrossRef]
- Chung, H.; Seo, S.; Moon, M.; Park, S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol. 2008, 198, 511–521. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Liu, Z.W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschop, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef]
- Lopez, N.E.; Gaston, L.; Lopez, K.R.; Coimbra, R.C.; Hageny, A.; Putnam, J.; Eliceiri, B.; Coimbra, R.; Bansal, V. Early ghrelin treatment attenuates disruption of the blood brain barrier and apoptosis after traumatic brain injury through a UCP-2 mechanism. Brain Res. 2012, 1489, 140–148. [Google Scholar]
- Hwang, S.; Moon, M.; Kim, S.; Hwang, L.; Ahn, K.J.; Park, S. Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response-4. Endocrine J. 2009, 56, 609–617. [Google Scholar] [CrossRef]
- Chung, H.; Chung, H.Y.; Bae, C.W.; Kim, C.J.; Park, S. Ghrelin suppresses tunicamycin- or thapsigargin-triggered endoplasmic reticulum stress-mediated apoptosis in primary cultured rat cortical neuronal cells. Endocrine J. 2011, 58, 409–420. [Google Scholar] [CrossRef]
- Wang, G.; Wang, W.; Zhao, J.; Ni, Y.; Zhou, X.; Zhang, W. Ghrelin prevents neuronal apoptosis and cognitive impairments in sepsis-associated encephalopathy. Neuroreport 2011, 22, 959–964. [Google Scholar] [CrossRef]
- Chung, H.; Kim, E.; Lee, D.H.; Seo, S.; Ju, S.; Lee, D.; Kim, H.; Park, S. Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 2007, 148, 148–159. [Google Scholar]
- Rezaeian, F.; Wettstein, R.; Scheuer, C.; Baumker, K.; Bachle, A.; Vollmar, B.; Menger, M.D.; Harder, Y. Ghrelin protects musculocutaneous tissue from ischemic necrosis by improving microvascular perfusion. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H603–H610. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.X.; Wu, D.; Wang, X.; Chen, H.L.; Chen, J.X.; Wang, X.X.; Wang, X.L.; Gan, L.; Guo, Z.Y.; Shi, G.X.; et al. Ghrelin protects against cobalt chloride-induced hypoxic injury in cardiac H9c2 cells by inhibiting oxidative stress and inducing autophagy. Peptides 2012, 38, 217–337. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, C.; Meng, B.; Tang, T.; Shi, Q.; Yang, H. Acute effect of ghrelin on ischemia/reperfusion injury in the rat spinal cord. Int. J. Mol. Sci. 2012, 13, 9864–9876. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef]
- Hallenbeck, J.M. Significance of the inflammatory response in brain ischemia. Acta Neurochir. Suppl. 1996, 66, 27–31. [Google Scholar]
- Wood, P.L. Microglia as a unique cellular target in the treatment of stroke: Potential neurotoxic mediators produced by activated microglia. Neurol. Res. 1995, 17, 242–248. [Google Scholar]
- Watanabe, H.; Abe, H.; Takeuchi, S.; Tanaka, R. Protective effect of microglial conditioning medium on neuronal damage induced by glutamate. Neurosci. Lett. 2000, 289, 53–56. [Google Scholar] [CrossRef]
- Wu, R.; Dong, W.; Zhou, M.; Cui, X.; Hank Simms, H.; Wang, P. Ghrelin improves tissue perfusion in severe sepsis via downregulation of endothelin-1. Cardiovasc. Res. 2005, 68, 318–326. [Google Scholar] [CrossRef]
- Li, Y.; Hai, J.; Li, L.; Chen, X.; Peng, H.; Cao, M.; Zhang, Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine 2012. [Google Scholar] [CrossRef]
- Sehirli, O.; Sener, E.; Sener, G.; Cetinel, S.; Erzik, C.; Yegen, B.C. Ghrelin improves burn-induced multiple organ injury by depressing neutrophil infiltration and the release of pro-inflammatory cytokines. Peptides 2008, 29, 1231–1240. [Google Scholar] [CrossRef]
- Qi, L.; Cui, X.; Dong, W.; Barrera, R.; Nicastro, J.; Coppa, G.F.; Wang, P.; Wu, R. Ghrelin attenuates brain injury after traumatic brain injury and uncontrolled hemorrhagic shock in rats. Mol. Med. 2012, 18, 186–193. [Google Scholar]
- Ersahin, M.; Toklu, H.Z.; Erzik, C.; Cetinel, S.; Akakin, D.; Velioglu-Ogunc, A.; Tetik, S.; Ozdemir, Z.N.; Sener, G.; Yegen, B.C. The anti-inflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats. J. Neurotrauma 2010, 27, 1143–1155. [Google Scholar] [CrossRef]
- Bansal, V.; Ryu, S.Y.; Lopez, N.; Allexan, S.; Krzyzaniak, M.; Eliceiri, B.; Baird, A.; Coimbra, R. Vagal stimulation modulates inflammation through a ghrelin mediated mechanism in traumatic brain injury. Inflammation 2012, 35, 214–220. [Google Scholar] [CrossRef]
- Broglio, F.; Benso, A.; Castiglioni, C.; Gottero, C.; Prodam, F.; Destefanis, S.; Gauna, C.; van der Lely, A.J.; Deghenghi, R.; Bo, M.; et al. The endocrine response to ghrelin as a function of gender in humans in young and elderly subjects. J. Clin. Endocrinol. Metab. 2003, 88, 1537–1542. [Google Scholar] [CrossRef]
- Aloi, J.A.; Gertz, B.J.; Hartman, M.L.; Huhn, W.C.; Pezzoli, S.S.; Wittreich, J.M.; Krupa, D.A.; Thorner, M.O. Neuroendocrine responses to a novel growth hormone secretagogue, L-692,429, in healthy older subjects. J. Clin. Endocrinol. Metab. 1994, 79, 943–949. [Google Scholar] [CrossRef]
- Yukawa, M.; Cummings, D.E.; Matthys, C.C.; Callahan, H.S.; Frayo, R.S.; Spiekerman, C.F.; Weigle, D.S. Effect of aging on the response of ghrelin to acute weight loss. J. Am. Geriatr. Soc. 2006, 54, 648–653. [Google Scholar] [CrossRef]
- Briggs, D.I.; Enriori, P.J.; Lemus, M.B.; Cowley, M.A.; Andrews, Z.B. Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 2010, 151, 4745–4755. [Google Scholar] [CrossRef]
- Tschop, M.; Weyer, C.; Tataranni, P.A.; Devanarayan, V.; Ravussin, E.; Heiman, M.L. Circulating ghrelin levels are decreased in human obesity. Diabetes 2001, 50, 707–709. [Google Scholar] [CrossRef]
- Poykko, S.M.; Kellokoski, E.; Horkko, S.; Kauma, H.; Kesaniemi, Y.A.; Ukkola, O. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 2003, 52, 2546–2553. [Google Scholar] [CrossRef]
- Poykko, S.; Ukkola, O.; Kauma, H.; Savolainen, M.J.; Kesaniemi, Y.A. Ghrelin Arg51Gln mutation is a risk factor for Type 2 diabetes and hypertension in a random sample of middle-aged subjects. Diabetologia 2003, 46, 455–458. [Google Scholar]
- Nogueiras, R.; Tovar, S.; Mitchell, S.E.; Rayner, D.V.; Archer, Z.A.; Dieguez, C.; Williams, L.M. Regulation of growth hormone secretagogue receptor gene expression in the arcuate nuclei of the rat by leptin and ghrelin. Diabetes 2004, 53, 2552–2558. [Google Scholar] [CrossRef]
- Akamizu, T.; Kangawa, K. Translational research on the clinical applications of ghrelin. Endocrine J. 2006, 53, 585–591. [Google Scholar] [CrossRef]
- Vestergaard, E.T.; Hansen, T.K.; Gormsen, L.C.; Jakobsen, P.; Moller, N.; Christiansen, J.S.; Jorgensen, J.O. Constant intravenous ghrelin infusion in healthy young men: Clinical pharmacokinetics and metabolic effects. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1829–E1836. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar]
- Jerlhag, E.; Egecioglu, E.; Dickson, S.L.; Engel, J.A. Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict. Biol. 2011, 16, 82–91. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar]
- Scherbakov, N.; Dirnagl, U.; Doehner, W. Body weight after stroke: Lessons from the obesity paradox. Stroke 2011, 42, 3646–3650. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Spencer, S.J.; Miller, A.A.; Andrews, Z.B. The Role of Ghrelin in Neuroprotection after Ischemic Brain Injury. Brain Sci. 2013, 3, 344-359. https://doi.org/10.3390/brainsci3010344
Spencer SJ, Miller AA, Andrews ZB. The Role of Ghrelin in Neuroprotection after Ischemic Brain Injury. Brain Sciences. 2013; 3(1):344-359. https://doi.org/10.3390/brainsci3010344
Chicago/Turabian StyleSpencer, Sarah J., Alyson A. Miller, and Zane B. Andrews. 2013. "The Role of Ghrelin in Neuroprotection after Ischemic Brain Injury" Brain Sciences 3, no. 1: 344-359. https://doi.org/10.3390/brainsci3010344
APA StyleSpencer, S. J., Miller, A. A., & Andrews, Z. B. (2013). The Role of Ghrelin in Neuroprotection after Ischemic Brain Injury. Brain Sciences, 3(1), 344-359. https://doi.org/10.3390/brainsci3010344