
Sleep and Risk of Multiple Sclerosis: Bridging the Gap Between Inflammation and Neurodegeneration via Glymphatic Failure

 , , and
, , and {kind=link}
Abstract
1. Introduction
2. Multiple Sclerosis Risk Factors
3. Sleep Physiological Functions in the Brain
4. Glymphatic System
5. Chronic Inflammation and Glymphatic Dysfunction in MS
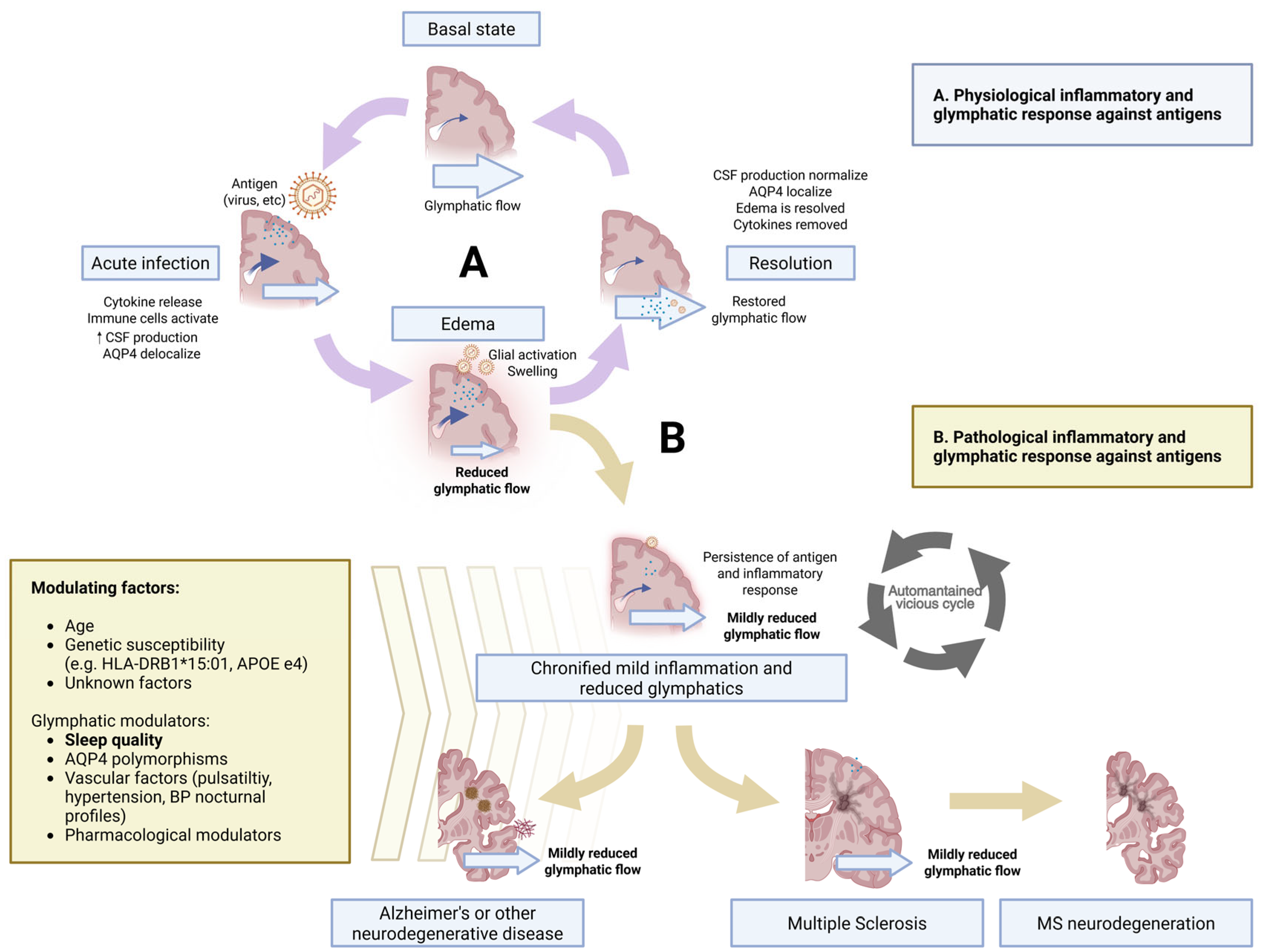
5.1. The Enduring Inflammation Model
5.2. Neuromyelitis Optica as a Prototype of Enduring Inflammation via Water-Channel Loss
6. Glymphatic Failure as the Bridge Between Inflammation and Neurodegeneration
6.1. Neurodegeneration in MS
6.2. Neurodegeneration in AD
6.3. Bridging the Gap: Glymphatic Modulators
7. Discussion and Future Directions
- Remove or silence the antigen. EBV vaccination, antiviral strategies, and broader pathogen-based prevention (e.g., shingles, influenza, pneumococcal vaccines) merit prospective trials that include imaging markers of glymphatic function.
- Ensure and restore deep sleep architecture. Behavioral sleep optimization, treatment of sleep-disordered breathing, and if needed, pharmacological consolidation with dual orexin-receptor antagonists such as lemborexant or daridorexant may be evaluated for their ability to boost slow-wave power and, in turn, glymphatic inflow. Strategies globally improving sleep quality may also positively impact the entire physiological process that supports memory consolidation.
- Normalize nocturnal haemodynamics. Although not so relevant in MS due to age of onset, controlling riser or non-dipping sleep blood-pressure patterns, particularly with blood–brain barrier-permeable β-blockers that also shrink astrocytic volume, could enhance perivascular pulsatility and widen interstitial space.
- Repolarize or augment AQP4 function. Small molecules that stabilize AQP4 at end-feet may help polarization and glymphatic flow.
- Non-pharmacological frequency-based interventions. Neuromodulation therapies enhancing glymphatic functioning, such as 40 Hz multisensory stimulation and others, may durably boost clearance and dampen inflammation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moutsianas, L.; Jostins, L.; Beecham, A.H.; Dilthey, A.T.; Xifara, D.K.; Ban, M.; Shah, T.S.; Patsopoulos, N.A.; Alfredsson, L.; Anderson, C.A.; et al. Class II HLA Interactions Modulate Genetic Risk for Multiple Sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Warner, H.B.; Carp, R.I. Multiple Sclerosis and Epstein-Barr Virus. Lancet 1981, 2, 1290. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Orr, N.; Steinman, L. Epstein-Barr Virus and the Immune Microenvironment in Multiple Sclerosis: Insights from High-Dimensional Brain Tissue Imaging. Proc. Natl. Acad. Sci. USA 2025, 122, e2425670122. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-Hydroxyvitamin D Levels and Risk of Multiple Sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Dobson, R.; Giovannoni, G.; Ramagopalan, S. V Smoking and Multiple Sclerosis: An Updated Meta-Analysis. PLoS ONE 2011, 6, e16149. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Marrodan, M. Multiple Sclerosis and Obesity: The Role of Adipokines. Front. Immunol. 2022, 13, 1038393. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Åkerstedt, T.; Olsson, T.; Alfredsson, L. Shift Work Influences Multiple Sclerosis Risk. Mult. Scler. 2015, 21, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, S.; Søndergaard, H.B.; Oturai, D.B.; Laursen, B.; Laursen, J.H.; Magyari, M.; Ullum, H.; Larsen, M.H.; Sellebjerg, F.; Oturai, A.B. Shift Work at Young Age Is Associated with Increased Risk of Multiple Sclerosis in a Danish Population. Mult. Scler. Relat. Disord. 2016, 9, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Åkerstedt, T.; Olsson, T.; Alfredsson, L.; Hedström, A.K. Insufficient Sleep during Adolescence and Risk of Multiple Sclerosis: Results from a Swedish Case-Control Study. J. Neurol. Neurosurg. Psychiatry 2023, 94, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Olsson, T.; Strid, P.; Kockum, I.; Alfredsson, L.; Hedström, A.K. Adolescent Sleep Patterns, Genetic Predisposition, and Risk of Multiple Sclerosis. Sleep 2024, 47, zsae156. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R. Sleep and Inflammation: Partners in Sickness and in Health. Nat. Rev. Immunol. 2019, 19, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Cirelli, C. Modulation of Brain Gene Expression during Sleep and Wakefulness: A Review of Recent Findings. Neuropsychopharmacology 2001, 25, S28–S35. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Nadel, L.; Payne, J.D. The Functions of Sleep: A Cognitive Neuroscience Perspective. Proc. Natl. Acad. Sci. USA 2022, 119, e2201795119. [Google Scholar] [CrossRef] [PubMed]
- Klinzing, J.G.; Niethard, N.; Born, J. Mechanisms of Systems Memory Consolidation during Sleep. Nat. Neurosci. 2019, 22, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, C.; Gutierrez, C.M.; Tononi, G. Extensive and Divergent Effects of Sleep and Wakefulness on Brain Gene Expression. Neuron 2004, 41, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Shockley, K.R.; Romer, M.A.; Galante, R.J.; Zimmerman, J.E.; Naidoo, N.; Baldwin, D.A.; Jensen, S.T.; Churchill, G.A.; Pack, A.I. Macromolecule Biosynthesis: A Key Function of Sleep. Physiol. Genom. 2007, 31, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Möller-Levet, C.S.; Archer, S.N.; Bucca, G.; Laing, E.E.; Slak, A.; Kabiljo, R.; Lo, J.C.Y.; Santhi, N.; von Schantz, M.; Smith, C.P.; et al. Effects of Insufficient Sleep on Circadian Rhythmicity and Expression Amplitude of the Human Blood Transcriptome. Proc. Natl. Acad. Sci. USA 2013, 110, E1132–E1141. [Google Scholar] [CrossRef] [PubMed]
- Mander, B.A. Local Sleep and Alzheimer’s Disease Pathophysiology. Front. Neurosci. 2020, 14, 525970. [Google Scholar] [CrossRef] [PubMed]
- Mander, B.A.; Dave, A.; Lui, K.K.; Sprecher, K.E.; Berisha, D.; Chappel-Farley, M.G.; Chen, I.Y.; Riedner, B.A.; Heston, M.; Suridjan, I.; et al. Inflammation, Tau Pathology, and Synaptic Integrity Associated with Sleep Spindles and Memory Prior to β-Amyloid Positivity. Sleep. 2022, 45, zsac135. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Guo, T.; Wang, Z.; Fang, Y.; Gu, L.; Cao, L.; Yang, W.; Gao, T.; Song, Z.; Tian, J.; et al. Neuroimaging Evidence of Glymphatic System Dysfunction in Possible REM Sleep Behavior Disorder and Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral Arterial Pulsation Drives Paravascular CSF-Interstitial Fluid Exchange in the Murine Brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Ringstad, G.; Valnes, L.M.; Dale, A.M.; Pripp, A.H.; Vatnehol, S.-A.S.; Emblem, K.E.; Mardal, K.-A.; Eide, P.K. Brain-Wide Glymphatic Enhancement and Clearance in Humans Assessed with MRI. JCI Insight 2018, 3, e121537. [Google Scholar] [CrossRef] [PubMed]
- Eide, P.K.; Pripp, A.H.; Berge, B.; Hrubos-Strøm, H.; Ringstad, G.; Valnes, L.M. Altered Glymphatic Enhancement of Cerebrospinal Fluid Tracer in Individuals with Chronic Poor Sleep Quality. J. Cereb. Blood Flow. Metab. 2022, 42, 1676–1692. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-Dependent Glymphatic Solute Transport in the Rodent Brain. Elife 2018, 7, e40070. [Google Scholar] [CrossRef] [PubMed]
- Brinker, T.; Stopa, E.; Morrison, J.; Klinge, P. A New Look at Cerebrospinal Fluid Circulation. Fluids Barriers CNS 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.P.; Gauberti, M.; Quenault, A.; Vivien, D.; Macrez, R.; Docagne, F. Reduced Spinal Cord Parenchymal Cerebrospinal Fluid Circulation in Experimental Autoimmune Encephalomyelitis. J. Cereb. Blood Flow. Metab. 2019, 39, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, X.; Wang, J.; Yang, L.; Zhuang, Y.; Dong, Y.; Tulupov, A.; Li, J.; Sun, J.; Li, J.; et al. Impaired Glymphatic System Function in Relapsing-Remitting Multiple Sclerosis: Insights from Diffusion Tensor Imaging along the Perivascular Space (DTI-ALPS). Neuroscience 2025, 580, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, A.; Cacciaguerra, L.; Pagani, E.; Preziosa, P.; Filippi, M.; Rocca, M.A. Glymphatic System Impairment in Multiple Sclerosis: Relation with Brain Damage and Disability. Brain 2022, 145, 2785–2795. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, Y.; Hagiwara, A.; Hoshino, Y.; Nakaya, M.; Kamagata, K.; Cossu, D.; Yokoyama, K.; Aoki, S.; Hattori, N. The Glymphatic System as a Potential Biomarker and Therapeutic Target in Secondary Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2024, 83, 105437. [Google Scholar] [CrossRef] [PubMed]
- Csomós, M.; Veréb, D.; Kocsis, K.; Faragó, P.; Tóth, E.; Antal, S.I.; Bozsik, B.; Tuka, B.; Király, A.; Szabó, N.; et al. Evaluation of the Glymphatic System in Relapsing Remitting Multiple Sclerosis by Measuring the Diffusion along the Perivascular Space. Magn. Reson. Imaging 2025, 117, 110319. [Google Scholar] [CrossRef] [PubMed]
- Granberg, T.; Moridi, T.; Brand, J.S.; Neumann, S.; Hlavica, M.; Piehl, F.; Ineichen, B. V Enlarged Perivascular Spaces in Multiple Sclerosis on Magnetic Resonance Imaging: A Systematic Review and Meta-Analysis. J. Neurol. 2020, 267, 3199–3212. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, F.L.-H.; Delle, C.; Nedergaard, M. The Glymphatic System (En)during Inflammation. Int. J. Mol. Sci. 2021, 22, 7491. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Burrows, S.R. Epstein-Barr Virus and Multiple Sclerosis: Potential Opportunities for Immunotherapy. Clin. Transl. Immunol. 2014, 3, e27. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed]
- Charcot, J.M. Leçons Sur Les Maladies Du Système Nerveux Faites à La Salpêtrière, 4th ed.; V. Adrien Delahaye: Paris, French, 1880; p. 2. [Google Scholar]
- Alghanimy, A.A.; Giovannoni, G.; Lechner-Scott, J.; Levy, M.; Yeh, E.A.; Hawkes, C.H. Is Multiple Sclerosis a Glymphaticopathy? Mult. Scler. Relat. Disord. 2023, 80, 105141. [Google Scholar] [CrossRef] [PubMed]
- Cullell, N.; Caruana, G.; Elias-Mas, A.; Delgado-Sanchez, A.; Artero, C.; Buongiorno, M.T.; Almería, M.; Ray, N.J.; Correa, S.A.L.; Krupinski, J. Glymphatic System Clearance and Alzheimer’s Disease Risk: A CSF Proteome-Wide Study. Alzheimers Res. Ther. 2025, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-X.; Zhang, W.-T.; Gao, S.-S.; Zhao, R.-Z.; Yu, W.-J.; Izquierdo, G. Sleep Disorders in Patients with Multiple Sclerosis in Spain. Neurologia 2021, 39, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Reider, N.; Cohen, J.; Trojano, M.; Sorensen, P.S.; Cutter, G.; Reingold, S.; Stuve, O. A Systematic Review of the Incidence and Prevalence of Sleep Disorders and Seizure Disorders in Multiple Sclerosis. Mult. Scler. 2015, 21, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Braley, T.J. Overview: A Framework for the Discussion of Sleep in Multiple Sclerosis. Curr. Sleep. Med. Rep. 2017, 3, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kotterba, S.; Neusser, T.; Norenberg, C.; Bussfeld, P.; Glaser, T.; Dörner, M.; Schürks, M. Sleep Quality, Daytime Sleepiness, Fatigue, and Quality of Life in Patients with Multiple Sclerosis Treated with Interferon Beta-1b: Results from a Prospective Observational Cohort Study. BMC Neurol. 2018, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Bahmani, D.; Esmaeili, L.; Shaygannejad, V.; Gerber, M.; Kesselring, J.; Lang, U.E.; Holsboer-Trachsler, E.; Brand, S. Stability of Mental Toughness, Sleep Disturbances, and Physical Activity in Patients with Multiple Sclerosis (MS)-A Longitudinal and Pilot Study. Front. Psychiatry 2018, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Sahraian, M.A.; Rezaali, S.; Hosseiny, M.; Doosti, R.; Tajik, A.; Naser Moghadasi, A. Sleep Disorder as a Triggering Factor for Relapse in Multiple Sclerosis. Eur. Neurol. 2017, 77, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG Marker of Optic-Spinal Multiple Sclerosis Binds to the Aquaporin-4 Water Channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Hinson, S.R.; Romero, M.F.; Popescu, B.F.G.; Lucchinetti, C.F.; Fryer, J.P.; Wolburg, H.; Fallier-Becker, P.; Noell, S.; Lennon, V.A. Molecular Outcomes of Neuromyelitis Optica (NMO)-IgG Binding to Aquaporin-4 in Astrocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Garton, T.; Gadani, S.P.; Gill, A.J.; Calabresi, P.A. Neurodegeneration and Demyelination in Multiple Sclerosis. Neuron 2024, 112, 3231–3251. [Google Scholar] [CrossRef] [PubMed]
- Pogoda-Wesołowska, A.; Dziedzic, A.; Maciak, K.; Stȩpień, A.; Dziaduch, M.; Saluk, J. Neurodegeneration and Its Potential Markers in the Diagnosing of Secondary Progressive Multiple Sclerosis. A Review. Front. Mol. Neurosci. 2023, 16, 1210091. [Google Scholar] [CrossRef] [PubMed]
- Valiukas, Z.; Tangalakis, K.; Apostolopoulos, V.; Feehan, J. Microglial Activation States and Their Implications for Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2025, 12, 100013. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking Immune Tolerance by Targeting Foxp3(+) Regulatory T Cells Mitigates Alzheimer’s Disease Pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and Protective Microglial Activation in Alzheimer’s Disease: A Prospective Study Using 18F-DPA-714 PET Imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef] [PubMed]
- Eyting, M.; Xie, M.; Michalik, F.; Heß, S.; Chung, S.; Geldsetzer, P. A Natural Experiment on the Effect of Herpes Zoster Vaccination on Dementia. Nature 2025, 641, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, H.; He, S.; Xia, T.; Chen, D.; Zhou, Y.; Liu, J.; Liu, M.; Sun, Z. Adult Vaccination as a Protective Factor for Dementia: A Meta-Analysis and Systematic Review of Population-Based Observational Studies. Front. Immunol. 2022, 13, 872542. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kwong, J.C.; Copes, R.; Hystad, P.; van Donkelaar, A.; Tu, K.; Brook, J.R.; Goldberg, M.S.; Martin, R.V.; Murray, B.J.; et al. Exposure to Ambient Air Pollution and the Incidence of Dementia: A Population-Based Cohort Study. Environ. Int. 2017, 108, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wilker, E.H.; Osman, M.; Weisskopf, M.G. Ambient Air Pollution and Clinical Dementia: Systematic Review and Meta-Analysis. BMJ 2023, 381, e071620. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Rábano, A.; Rodal, I.; Carrasco, L. Different Brain Regions Are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef] [PubMed]
- Cottrill, R.; Ekanayake, A.; Grove, C.; Peiris, S.; Corbett, N.; Ahmed, B.; Jens, W.; Brearly, T.; Kanekar, S.; Eslinger, P.; et al. Alzheimer’s Disease (AD) in Multiple Sclerosis (MS): A Systematic Review of Published Cases, Mechanistic Links between AD and MS, and Possible Clinical Evaluation of AD in MS. J. Alzheimers Dis. Rep. 2025, 9, 25424823251316136. [Google Scholar] [CrossRef] [PubMed]
- Novarella, F.; Nicolella, V.; Fiorenza, M.; Falco, F.; Monteiro, I.; Corsini, G.; Ranucci, D.; Carotenuto, A.; Petracca, M.; Lanzillo, R.; et al. Neurofilament Light Chain and Alzheimer Pathology Biomarkers in Elderly People with Multiple Sclerosis. J. Neurol. Sci. 2025, 475, 123562. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic Variation in Aquaporin-4 Moderates the Relationship between Sleep and Brain Aβ-Amyloid Burden. Transl. Psychiatry 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, Q.; Luo, X.; Hong, H.; Fang, Y.; Xie, L.; Zhang, Y.; Lin, M.; Wang, S.; Li, K.; et al. Association Between Single Nucleotide Polymorphisms in the Aquaporin-4 Gene and Longitudinal Changes in White Matter Free Water and Cognitive Function in Non-Demented Older Adults. Hum. Brain Mapp. 2025, 46, e70171. [Google Scholar] [CrossRef] [PubMed]
- Fultz, N.E.; Bonmassar, G.; Setsompop, K.; Stickgold, R.A.; Rosen, B.R.; Polimeni, J.R.; Lewis, L.D. Coupled Electrophysiological, Hemodynamic, and Cerebrospinal Fluid Oscillations in Human Sleep. Science 2019, 366, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Jiang-Xie, L.-F.; Drieu, A.; Bhasiin, K.; Quintero, D.; Smirnov, I.; Kipnis, J. Neuronal Dynamics Direct Cerebrospinal Fluid Perfusion and Brain Clearance. Nature 2024, 627, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Buongiorno, M.; Sánchez-Benavides, G.; Caruana, G.; Elias-Mas, A.; Artero, C.; Cullell, N.; Delgado, P.; Giraldo, D.M.; Marzal-Espí, C.; Grau-Rivera, O.; et al. Abnormal Sleep Blood Pressure Patterns Are Associated with Diffusion Tensor Imaging along Perivascular Spaces Index in Cognitively Impaired Individuals. medRxiv 2025. [Google Scholar] [CrossRef]
- Mestre, H.; Tithof, J.; Du, T.; Song, W.; Peng, W.; Sweeney, A.M.; Olveda, G.; Thomas, J.H.; Nedergaard, M.; Kelley, D.H. Flow of Cerebrospinal Fluid Is Driven by Arterial Pulsations and Is Reduced in Hypertension. Nat. Commun. 2018, 9, 4878. [Google Scholar] [CrossRef] [PubMed]
- Buongiorno, M.; Sánchez-Benavides, G.; Marzal-Espí, C.; Giraldo, D.M.; Krupinski, J.; Cullell, N.; Grau-Rivera, O.; Suárez-Calvet, M.; Gispert, J.D.; de la Sierra, A. Blood-Brain Barrier Permeable β-Blockers Association with Alzheimer’s Disease Cerebrospinal Fluid Biomarkers Levels in Non-Demented Individuals. J. Alzheimer’s Dis. 2024, 102, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, T.; Djonlagic, I.; Dauvilliers, Y.; Sadeghi, K.; Little, D.; Datta, A.N.; Hubbard, J.; Hajak, G.; Krystal, A.; Olivieri, A.; et al. Effect of Daridorexant on Sleep Architecture in Patients with Chronic Insomnia Disorder: A Pooled Post Hoc Analysis of Two Randomized Phase 3 Clinical Studies. Sleep. 2024, 47, zsae098. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.; Murphy, P.; Zammit, G.; Mayleben, D.; Kumar, D.; Dhadda, S.; Filippov, G.; LoPresti, A.; Moline, M. Comparison of Lemborexant with Placebo and Zolpidem Tartrate Extended Release for the Treatment of Older Adults With Insomnia Disorder: A Phase 3 Randomized Clinical Trial. JAMA Netw. Open 2019, 2, e1918254. [Google Scholar] [CrossRef] [PubMed]
- Murdock, M.H.; Yang, C.-Y.; Sun, N.; Pao, P.-C.; Blanco-Duque, C.; Kahn, M.C.; Kim, T.; Lavoie, N.S.; Victor, M.B.; Islam, M.R.; et al. Multisensory Gamma Stimulation Promotes Glymphatic Clearance of Amyloid. Nature 2024, 627, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hainke, L.; Dowsett, J.; Spitschan, M.; Priller, J. 40 Hz Visual Stimulation during Sleep Evokes Neuronal Gamma Activity in NREM and REM Stages. Sleep. 2025, 48, zsae299. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buongiorno, M.; Tur, C.; Giraldo, D.M.; Cullell, N.; Krupinski, J.; Lanzillo, R.; Sánchez-Benavides, G. Sleep and Risk of Multiple Sclerosis: Bridging the Gap Between Inflammation and Neurodegeneration via Glymphatic Failure. Brain Sci. 2025, 15, 766. https://doi.org/10.3390/brainsci15070766
Buongiorno M, Tur C, Giraldo DM, Cullell N, Krupinski J, Lanzillo R, Sánchez-Benavides G. Sleep and Risk of Multiple Sclerosis: Bridging the Gap Between Inflammation and Neurodegeneration via Glymphatic Failure. Brain Sciences. 2025; 15(7):766. https://doi.org/10.3390/brainsci15070766
Chicago/Turabian StyleBuongiorno, Mariateresa, Carmen Tur, Darly Milena Giraldo, Natalia Cullell, Jerzy Krupinski, Roberta Lanzillo, and Gonzalo Sánchez-Benavides. 2025. "Sleep and Risk of Multiple Sclerosis: Bridging the Gap Between Inflammation and Neurodegeneration via Glymphatic Failure" Brain Sciences 15, no. 7: 766. https://doi.org/10.3390/brainsci15070766
APA StyleBuongiorno, M., Tur, C., Giraldo, D. M., Cullell, N., Krupinski, J., Lanzillo, R., & Sánchez-Benavides, G. (2025). Sleep and Risk of Multiple Sclerosis: Bridging the Gap Between Inflammation and Neurodegeneration via Glymphatic Failure. Brain Sciences, 15(7), 766. https://doi.org/10.3390/brainsci15070766