

The Role of Immune Cells in Moyamoya Disease
 , ,
, , {kind=link}
{kind=link}
Abstract
1. Introduction
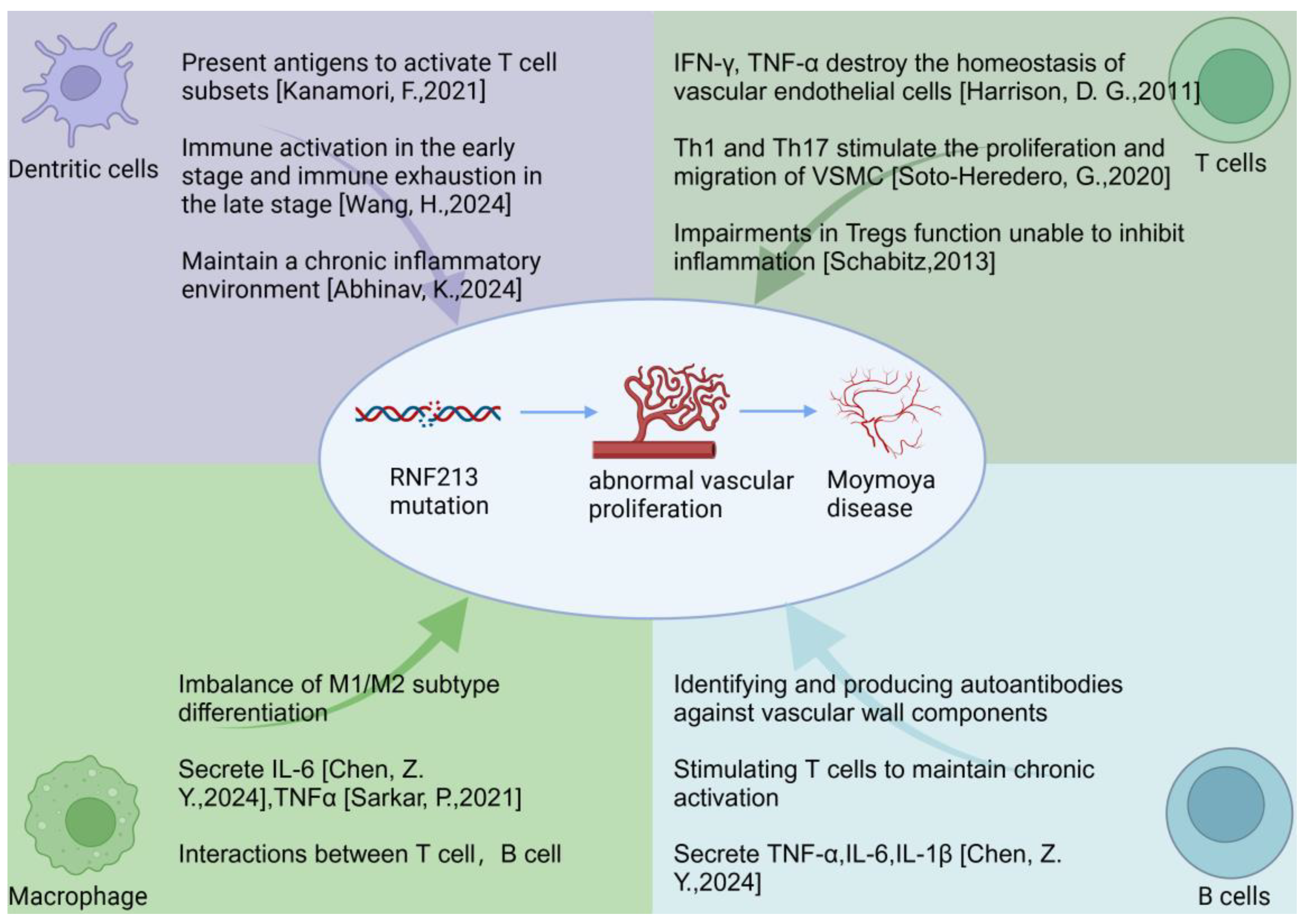
2. Overview of Immune Cells Involved in MMD
2.1. T Cells
2.1.1. T Cells and Vascular Aging
2.1.2. T Cells and Vascular Hypertension
2.1.3. T Cells and Vascular Inflammation
2.1.4. T Cell and MMD: Histopathological Evidence
2.1.5. T Cell and MMD: Peripheral Immune Profiles
2.2. B Cells
2.2.1. Autoantibodies and Vascular Diseases
2.2.2. B Cells and Vascular Inflammation
2.2.3. B Cells and Immune Dysregulation in MMD
2.2.4. Autoimmune Responses and B Cell Involvement
2.3. Macrophage
2.3.1. Macrophage and Vascular Inflammation
2.3.2. Macrophage Polarization in MMD
2.3.3. Macrophage Infiltration and Vascular Remodeling
2.3.4. Macrophage and Endothelial Dysfunction
2.4. Dendritic Cells
2.4.1. Dendritic Cells and Vascular Inflammation
2.4.2. Dendritic Cells in Inflammation and Vascular Remodeling in MMD
2.4.3. Dendritic Cells and Immune Dysregulation in MMD
2.5. Others
2.6. Therapeutic Implications
2.7. Challenges and Future Directions
3. Potential Mechanisms Linking Immune Cells to MMD Pathogenesis
3.1. Chronic Inflammation
3.2. Endothelial Damage
3.3. Smooth Muscle Cell Proliferation
3.4. Autoimmunity
3.5. Cytokine-Mediated Effects
3.6. Dysregulation of Angiogenic Factors
3.7. Function of RNF213 Gene
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, D.; Huang, L.; Huang, Z.; Zhou, Q.; Yang, X.; Gu, H.; Li, Z.; Shi, Y.; Gan, L.; Wang, H.; et al. Epidemiology of moyamoya disease in china: A nationwide hospital-based study. Lancet Reg. Health West Pac. 2022, 18, 100331. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y.; Hattori, Y.; Liu, W.; Kobayashi, H.; Ishiyama, H.; Yoshimoto, T.; Miyawaki, S.; Clausen, T.; Bang, O.Y.; et al. Moyamoya disease: Diagnosis and interventions. Lancet Neurol. 2022, 21, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Fujimura, M.; Takahashi, J.; Kataoka, H.; Ogasawara, K.; Iwama, T.; Tominaga, T.; Miyamoto, S. Research Committee on Moyamoya Disease of the Ministry of Health L, Welfare J. Diagnostic Criteria for Moyamoya Disease—2021 Revised Version. Neurol. Med. Chir. 2022, 62, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Mineharu, Y.; Miyamoto, S. RNF213 and GUCY1A3 in Moyamoya Disease: Key Regulators of Metabolism, Inflammation, and Vascular Stability. Front. Neurol. 2021, 12, 687088. [Google Scholar] [CrossRef]
- Zhang, L.; Rashad, S.; Zhou, Y.; Niizuma, K.; Tominaga, T. RNF213 loss of function reshapes vascular transcriptome and spliceosome leading to disrupted angiogenesis and aggravated vascular inflammatory responses. J. Cereb. Blood Flow Metab. 2022, 42, 2107–2122. [Google Scholar] [CrossRef]
- Ikeda, E. Systemic vascular changes in spontaneous occlusion of the circle of Willis. Stroke 1991, 22, 1358–1362. [Google Scholar] [CrossRef]
- Morimoto, T.; Mineharu, Y.; Ono, K.; Nakatochi, M.; Ichihara, S.; Kabata, R.; Takagi, Y.; Cao, Y.; Zhao, L.; Kobayashi, H.; et al. Significant association of RNF213 p.R4810K, a moyamoya susceptibility variant, with coronary artery disease. PLoS ONE 2017, 12, e0175649. [Google Scholar] [CrossRef]
- Koyama, S.; Ito, K.; Terao, C.; Akiyama, M.; Horikoshi, M.; Momozawa, Y.; Matsunaga, H.; Ieki, H.; Ozaki, K.; Onouchi, Y.; et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020, 52, 1169–1177. [Google Scholar] [CrossRef]
- Tian, H.; Yu, K.; He, L.; Xu, H.; Han, C.; Zhang, X.; Wang, X.; Zhang, X.; Zhang, L.; Gao, G.; et al. RNF213 modulates gamma-herpesvirus infection and reactivation via targeting the viral Replication and Transcription Activator. Proc. Natl. Acad. Sci. USA 2023, 120, e2218825120. [Google Scholar] [CrossRef]
- Otten, E.G.; Werner, E.; Crespillo-Casado, A.; Boyle, K.B.; Dharamdasani, V.; Pathe, C.; Santhanam, B.; Randow, F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Guo, Z.N.; Shi, M.; Yang, Y.; Rao, M. Etiology and pathogenesis of Moyamoya Disease: An update on disease prevalence. Int. J. Stroke 2017, 12, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Pollaci, G.; Gorla, G.; Potenza, A.; Carrozzini, T.; Canavero, I.; Bersano, A.; Gatti, L. Novel Multifaceted Roles for RNF213 Protein. Int. J. Mol. Sci. 2022, 23, 4492. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, X.; Ni, J. RNF213 in moyamoya disease: Genotype-phenotype association and the underlying mechanism. Chin. Med. J. 2024, 137, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Asselman, C.; Hemelsoet, D.; Eggermont, D.; Dermaut, B.; Impens, F. Moyamoya disease emerging as an immune-related angiopathy. Trends Mol. Med. 2022, 28, 939–950. [Google Scholar] [CrossRef]
- Ge, P.; Tao, C.; Wang, W.; He, Q.; Liu, C.; Zheng, Z.; Mou, S.; Zhang, B.; Liu, X.; Zhang, Q.; et al. Circulating immune cell landscape and T-cell abnormalities in patients with moyamoya disease. Clin. Transl. Med. 2024, 14, e1647. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, Y.; Zhang, J.; Liu, Z.; Hao, X.; Wang, X.; He, S.; Wang, R. Characterization of PANoptosis-related genes and the immune landscape in moyamoya disease. Sci. Rep. 2024, 14, 10278. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, X.; Sheng, J.; Fu, Y.; Nie, D.; You, X.; Chen, Y.; Yang, X.; Ling, Q.; Zhang, H.; et al. RNF213 promotes Treg cell differentiation by facilitating K63-linked ubiquitination and nuclear translocation of FOXO1. Nat. Commun. 2024, 15, 5961. [Google Scholar] [CrossRef]
- Liu, E.; Liu, C.; Jin, L.; Zhou, H.; Tan, X.; Zhang, G.; Tao, W.; Gao, X.; Zhao, H.; Luo, C.; et al. Clinical value of the systemic immune-inflammation index in moyamoya disease. Front. Neurol. 2023, 14, 1123951. [Google Scholar] [CrossRef]
- Jin, F.; Duan, C. Identification of immune-infiltrated hub genes as potential biomarkers of Moyamoya disease by bioinformatics analysis. Orphanet J. Rare Dis. 2022, 17, 80. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Fukai, N.; Aoyagi, M.; Yamamoto, M.; Sakamoto, H.; Ogami, K.; Matsushima, Y.; Yamamoto, K. Human arterial smooth muscle cell strains derived from patients with moyamoya disease: Changes in biological characteristics and proliferative response during cellular aging in vitro. Mech. Ageing Dev. 1994, 75, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Ord, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Immune modulation of resistance artery remodelling. Basic Clin. Pharmacol. Toxicol. 2012, 110, 70–72. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal. Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 2021, 1, 598–615. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef]
- Schmitt, V.; Rink, L.; Uciechowski, P. The Th17/Treg balance is disturbed during aging. Exp. Gerontol. 2013, 48, 1379–1386. [Google Scholar] [CrossRef]
- Shirakawa, K.; Sano, M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells 2021, 10, 2435. [Google Scholar] [CrossRef]
- Antonicelli, F.; Bellon, G.; Debelle, L.; Hornebeck, W. Elastin-elastases and inflamm-aging. Curr. Top Dev. Biol. 2007, 79, 99–155. [Google Scholar] [CrossRef]
- Cao, H.; Diao, J.; Liu, H.; Liu, S.; Liu, J.; Yuan, J.; Lin, J. The Pathogenicity and Synergistic Action of Th1 and Th17 Cells in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2023, 29, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Hu, L.; Jiang, F. Study of cytokine-induced immunity in bullous pemphigoid: Recent developments. Ann. Med. 2023, 55, 2280991. [Google Scholar] [CrossRef]
- Jin, K.; Parreau, S.; Warrington, K.J.; Koster, M.J.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Regulatory T Cells in Autoimmune Vasculitis. Front. Immunol. 2022, 13, 844300. [Google Scholar] [CrossRef]
- Ohkura, N.; Sakaguchi, S. Transcriptional and epigenetic basis of Treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Frasca, D.; Saada, Y.B.; Garcia, D.; Friguet, B. Effects of cellular senescence on metabolic pathways in non-immune and immune cells. Mech. Ageing Dev. 2021, 194, 111428. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Madhur, M.S.; Elijovich, F.; Alexander, M.R.; Pitzer, A.; Ishimwe, J.; Van Beusecum, J.P.; Patrick, D.M.; Smart, C.D.; Kleyman, T.R.; Kingery, J.; et al. Hypertension: Do Inflammation and Immunity Hold the Key to Solving this Epidemic? Circ. Res. 2021, 128, 908–933. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L. Immune mechanisms in hypertension and vascular injury. Clin. Sci. (Lond.) 2014, 126, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Guzik, T.J.; Goronzy, J.; Weyand, C. Is hypertension an immunologic disease? Curr. Cardiol. Rep. 2008, 10, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Idris-Khodja, N.; Mian, M.O.; Paradis, P.; Schiffrin, E.L. Dual opposing roles of adaptive immunity in hypertension. Eur. Heart J. 2014, 35, 1238–1244. [Google Scholar] [CrossRef]
- Marko, L.; Kvakan, H.; Park, J.K.; Qadri, F.; Spallek, B.; Binger, K.J.; Bowman, E.P.; Kleinewietfeld, M.; Fokuhl, V.; Dechend, R.; et al. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 2012, 60, 1430–1436. [Google Scholar] [CrossRef]
- Satou, R.; Miyata, K.; Gonzalez-Villalobos, R.A.; Ingelfinger, J.R.; Navar, L.G.; Kobori, H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J. 2012, 26, 1821–1830. [Google Scholar] [CrossRef]
- Wenzel, U.O.; Bode, M.; Kurts, C.; Ehmke, H. Salt, inflammation, IL-17 and hypertension. Br. J. Pharmacol. 2019, 176, 1853–1863. [Google Scholar] [CrossRef]
- Guzik, T.J.; Nosalski, R.; Maffia, P.; Drummond, G.R. Immune and inflammatory mechanisms in hypertension. Nat. Rev. Cardiol. 2024, 21, 396–416. [Google Scholar] [CrossRef]
- Imiela, A.M.; Mikolajczyk, T.P.; Siedlinski, M.; Dobrowolski, P.; Konior-Rozlachowska, A.; Wrobel, A.; Biernat-Kaluza, E.; Januszewicz, M.; Guzik, B.; Guzik, T.J.; et al. Th17/Treg imbalance in patients with primary hyperaldosteronism and resistant hypertension. Pol. Arch. Intern. Med. 2022, 132, 132. [Google Scholar] [CrossRef]
- Meng, X.; Yang, J.; Dong, M.; Zhang, K.; Tu, E.; Gao, Q.; Chen, W.; Zhang, C.; Zhang, Y. Regulatory T cells in cardiovascular diseases. Nat. Rev. Cardiol. 2016, 13, 167–179. [Google Scholar] [CrossRef]
- Ferro, C.J.; Webb, D.J. Endothelial dysfunction and hypertension. Drugs 1997, 53 (Suppl. 1), 30–41. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H. Endothelial dysfunction and hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef] [PubMed]
- Feihl, F.; Liaudet, L.; Levy, B.I.; Waeber, B. Hypertension and microvascular remodelling. Cardiovasc. Res. 2008, 78, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (raas): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Ngwenyama, N.; Salvador, A.M.; Velazquez, F.; Nevers, T.; Levy, A.; Aronovitz, M.; Luster, A.D.; Huggins, G.S.; Alcaide, P. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight 2019, 4, e125527. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef]
- Shao, J.; Nangaku, M.; Miyata, T.; Inagi, R.; Yamada, K.; Kurokawa, K.; Fujita, T. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 2003, 42, 31–38. [Google Scholar] [CrossRef]
- Masuda, J.; Ogata, J.; Yutani, C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in moyamoya disease. Stroke 1993, 24, 1960–1967. [Google Scholar] [CrossRef]
- Li, S.; Han, Y.; Zhang, Q.; Tang, D.; Li, J.; Weng, L. Comprehensive molecular analyses of an autoimmune-related gene predictive model and immune infiltrations using machine learning methods in moyamoya disease. Front. Mol. Biosci. 2022, 9, 991425. [Google Scholar] [CrossRef]
- Cao, L.; Ai, Y.; Dong, Y.; Li, D.; Wang, H.; Sun, K.; Wang, C.; Zhang, M.; Yan, D.; Li, H.; et al. Bioinformatics analysis reveals the landscape of immune cell infiltration and novel immune-related biomarkers in moyamoya disease. Front. Genet. 2023, 14, 1101612. [Google Scholar] [CrossRef] [PubMed]
- Schabitz, W.R. Regulatory T cells in ischemic stroke: Helpful or hazardous? Stroke 2013, 44, e84. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Q.N.; Li, J.; Liu, S.; Wang, X.; Yu, D.; Zou, Z.X.; Gao, G.; Zhang, Q.; Hao, F.B.; et al. Proteomic and metabolomic characterizations of moyamoya disease patient sera. Brain Behav. 2023, 13, e3328. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; De Rosa, V.; Procaccini, C.; Matarese, G. Regulatory T cells, leptin and angiogenesis. Chem. Immunol. Allergy 2014, 99, 155–169. [Google Scholar] [CrossRef]
- Kanamori, F.; Yokoyama, K.; Ota, A.; Yoshikawa, K.; Karnan, S.; Maruwaka, M.; Shimizu, K.; Ota, S.; Uda, K.; Araki, Y.; et al. Transcriptome-wide analysis of intracranial artery in patients with moyamoya disease showing upregulation of immune response, and downregulation of oxidative phosphorylation and DNA repair. Neurosurg. Focus 2021, 51, E3. [Google Scholar] [CrossRef]
- Wang, H.; Hao, F.; Feng, J.; Zhang, Q.; Zhang, Z.; Li, B.; Zhang, H.; Yu, X.; Han, C.; Duan, L. Clinical Course, Therapy, and Long-Term Outcomes of Children with Moyamoya Disease and Posterior Cerebral Artery Involvement. Neurology 2024, 103, e209658. [Google Scholar] [CrossRef]
- Abhinav, K.; Lee, A.G.; Pendharkar, A.V.; Bigder, M.; Bet, A.; Rosenberg-Hasson, Y.; Cheng, M.Y.; Steinberg, G.K. Comprehensive Profiling of Secreted Factors in the Cerebrospinal Fluid of Moyamoya Disease Patients. Transl. Stroke Res. 2024, 15, 399–408. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Yu, X.Q.; Xiang, Y.Y.; Liu, L.H.; Yin, X.P. Moyamoya syndrome may result from psoriasis: Four case reports. World J. Clin. Cases 2024, 12, 1830–1836. [Google Scholar] [CrossRef]
- Sarkar, P.; Thirumurugan, K. New insights into TNFalpha/PTP1B and PPARgamma pathway through RNF213- a link between inflammation, obesity, insulin resistance, and Moyamoya disease. Gene 2021, 771, 145340. [Google Scholar] [CrossRef]
- Carbone, F.; Montecucco, F. Inflammation in arterial diseases. IUBMB Life 2015, 67, 18–28. [Google Scholar] [CrossRef]
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Guggenberger, K.; Krafft, A.J.; Ludwig, U.; Raithel, E.; Forman, C.; Meckel, S.; Hennig, J.; Bley, T.A.; Vogel, P. Intracranial vessel wall imaging framework—Data acquisition, processing, and visualization. Magn. Reson. Imaging 2021, 83, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Arslan, S.; Korkmazer, B.; Kizilkilic, O. Intracranial vessel wall imaging. Curr. Opin. Rheumatol. 2021, 33, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef]
- Tektonidou, M.G. Cardiovascular disease risk in antiphospholipid syndrome: Thrombo-inflammation and atherothrombosis. J. Autoimmun. 2022, 128, 102813. [Google Scholar] [CrossRef]
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532. [Google Scholar] [CrossRef]
- Emathinger, J.M.; Nelson, J.W.; Gurley, S.B. Advances in use of mouse models to study the renin-angiotensin system. Mol. Cell Endocrinol. 2021, 529, 111255. [Google Scholar] [CrossRef]
- Kinoshita, O.; Kawano, Y.; Yoshimi, H.; Ashida, T.; Yoshida, K.; Akabane, S.; Kuramochi, M.; Omae, T. Acute and chronic effects of anti-endothelin-1 antibody on blood pressure in spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 1991, 17 (Suppl. 7), S511–S513. [Google Scholar] [CrossRef]
- Massicotte-Azarniouch, D.; Herrera, C.A.; Jennette, J.C.; Falk, R.J.; Free, M.E. Mechanisms of vascular damage in ANCA vasculitis. Semin. Immunopathol. 2022, 44, 325–345. [Google Scholar] [CrossRef]
- Knight, J.S.; Kanthi, Y. Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin. Immunopathol. 2022, 44, 347–362. [Google Scholar] [CrossRef]
- Siklova, M.; Koc, M.; Rossmeislova, L.; Kraml, P. Serum oxldl-beta2gpi complex reflects metabolic syndrome and inflammation in adipose tissue in obese. Int. J. Obes. (Lond.) 2018, 42, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Baudin, B. Polymorphism in angiotensin ii receptor genes and hypertension. Exp. Physiol. 2005, 90, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, S.; Baltaro, R.; Hurley, J. Systemic vasculitis. Am. Fam. Physician 2011, 83, 556–565. [Google Scholar] [PubMed]
- Kyaw, T.; Tay, C.; Krishnamurthi, S.; Kanellakis, P.; Agrotis, A.; Tipping, P.; Bobik, A.; Toh, B.H. B1a b lymphocytes are atheroprotective by secreting natural igm that increases igm deposits and reduces necrotic cores in atherosclerotic lesions. Circ. Res. 2011, 109, 830–840. [Google Scholar] [CrossRef]
- Hosseini, H.; Li, Y.; Kanellakis, P.; Tay, C.; Cao, A.; Liu, E.; Peter, K.; Tipping, P.; Toh, B.H.; Bobik, A.; et al. Toll-Like Receptor (TLR)4 and MyD88 are Essential for Atheroprotection by Peritoneal B1a B Cells. J. Am. Heart Assoc. 2016, 5, e002947. [Google Scholar] [CrossRef]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef]
- Tian, J.; Zekzer, D.; Hanssen, L.; Lu, Y.; Olcott, A.; Kaufman, D.L. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J. Immunol. 2001, 167, 1081–1089. [Google Scholar] [CrossRef]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Mejia-Munne, J.C.; Ellis, J.A.; Feldstein, N.A.; Meyers, P.M.; Connolly, E.S. Moyamoya and Inflammation. World Neurosurg. 2017, 100, 575–578. [Google Scholar] [CrossRef]
- Thamamongood, T.; Hara, S.; Akagawa, H.; Inaji, M.; Tanaka, Y.; Nariai, T.; Maehara, T. Synergistic Interaction of Thyroid Autoantibodies and Ring Finger Protein 213 Variant in Moyamoya Disease. Neurol. Med. Chir. 2024, 64, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, X.; Chen, Z.; Liebeskind, D.S.; Lou, M. Elevated thyroid autoantibodies and intracranial stenosis in stroke at an early age. Int. J. Stroke 2014, 9, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflug. Arch. 2017, 469, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in Inflammatory Bowel Disease. Int. J. Inflam. 2015, 2015, 970890. [Google Scholar] [CrossRef] [PubMed]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Fujimura, T.; Kakizaki, A.; Sato-Maeda, M.; Niizuma, K.; Tomata, Y.; Aiba, S.; Tominaga, T. Increased serum production of soluble CD163 and CXCL5 in patients with moyamoya disease: Involvement of intrinsic immune reaction in its pathogenesis. Brain Res. 2018, 1679, 39–44. [Google Scholar] [CrossRef]
- Nagata, E.; Masuda, H.; Nakayama, T.; Netsu, S.; Yuzawa, H.; Fujii, N.; Kohara, S.; Sorimachi, T.; Osada, T.; Imazeki, R.; et al. Insufficient production of IL-10 from M2 macrophages impairs in vitro endothelial progenitor cell differentiation in patients with Moyamoya disease. Sci. Rep. 2019, 9, 16752. [Google Scholar] [CrossRef]
- Dixon, K.B.; Davies, S.S.; Kirabo, A. Dendritic cells and isolevuglandins in immunity, inflammation, and hypertension. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H368–H374. [Google Scholar] [CrossRef]
- Tashiro, R.; Niizuma, K.; Kasamatsu, J.; Okuyama, Y.; Rashad, S.; Kikuchi, A.; Fujimura, M.; Kure, S.; Ishii, N.; Tominaga, T. Dysregulation of Rnf 213 gene contributes to T cell response via antigen uptake, processing, and presentation. J. Cell Physiol. 2021, 236, 7554–7564. [Google Scholar] [CrossRef]
- Liu, C.; Ge, P.; Zhang, B.; Chan, L.; Pang, Y.; Tao, C.; Li, J.; He, Q.; Liu, W.; Mou, S.; et al. Mass cytometry revealed the circulating immune cell landscape across different Suzuki stages of Moyamoya disease. Immunol. Res. 2024, 72, 654–664. [Google Scholar] [CrossRef]
- Shirozu, N.; Ohgidani, M.; Hata, N.; Tanaka, S.; Inamine, S.; Sagata, N.; Kimura, T.; Inoue, I.; Arimura, K.; Nakamizo, A.; et al. Angiogenic and inflammatory responses in human induced microglia-like (iMG) cells from patients with Moyamoya disease. Sci. Rep. 2023, 13, 14842. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Wang, J.; Zhang, H.; Hao, F.B.; Gao, G.; Liu, S.M.; Wang, X.P.; Li, J.J.; Zou, Z.X.; Guo, Q.B.; et al. High Level of Serum Complement C3 Expression is Associated with Postoperative Vasculopathy Progression in Moyamoya Disease. J. Inflamm. Res. 2024, 17, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, X.; Gu, X.; Dong, M.; Huang, L.; Mao, C.; Xia, S.; Yang, H.; Bao, X.; Yang, Y.; et al. GM-CSF Promotes the Development of Dysfunctional Vascular Networks in Moyamoya Disease. Neurosci. Bull. 2024, 40, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Gao, J.; Liu, J.; Wang, C.; Li, A.; Wang, J. Study on novel nanoparticle slow-release drugs for moyamoya disease. J. Nanosci. Nanotechnol. 2021, 21, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kazumata, K.; Nakatani, E.; Houkin, K.; Kanatani, Y. Characteristics of moyamoya disease based on national registry data in japan. Stroke 2019, 50, 1973–1980. [Google Scholar] [CrossRef]
- Ando, S.; Tsutsui, S.; Miyoshi, K.; Sato, S.; Yanagihara, W.; Setta, K.; Chiba, T.; Fujiwara, S.; Kobayashi, M.; Yoshida, K.; et al. Cilostazol may improve cognition better than clopidogrel in non-surgical adult patients with ischemic moyamoya disease: Subanalysis of a prospective cohort. Neurol. Res. 2019, 41, 480–487. [Google Scholar] [CrossRef]
- Liu, Z.; He, S.; Xu, Z.; Duan, R.; Yuan, L.; Xiao, C.; Yi, Z.; Wang, R. Association between white matter impairment and cognitive dysfunction in patients with ischemic moyamoya disease. BMC Neurol. 2020, 20, 302. [Google Scholar] [CrossRef]
- Farooq, M.U.; Min, J.; Goshgarian, C.; Gorelick, P.B. Pharmacotherapy for vascular cognitive impairment. CNS Drugs 2017, 31, 759–776. [Google Scholar] [CrossRef]
- Porras, J.L.; Yang, W.; Xu, R.; Garzon-Muvdi, T.; Caplan, J.M.; Colby, G.P.; Coon, A.L.; Ahn, E.S.; Tamargo, R.J.; Huang, J. Effectiveness of ipsilateral stroke prevention between conservative management and indirect revascularization for moyamoya disease in a north american cohort. World Neurosurg. 2018, 110, e928–e936. [Google Scholar] [CrossRef]
- Ito, M.; Kawabori, M.; Sugiyama, T.; Tokairin, K.; Tatezawa, R.; Uchino, H.; Kazumata, K.; Houkin, K.; Fujimura, M. Impact of rnf213 founder polymorphism (p.R4810k) on the postoperative development of indirect pial synangiosis after direct/indirect combined revascularization surgery for adult moyamoya disease. Neurosurg. Rev. 2022, 45, 2305–2313. [Google Scholar] [CrossRef]
- Shin, H.S.; Park, G.H.; Choi, E.S.; Park, S.Y.; Kim, D.S.; Chang, J.; Hong, J.M. Rnf213 variant and autophagic impairment: A pivotal link to endothelial dysfunction in moyamoya disease. J. Cereb. Blood Flow Metab. 2024, 44, 1801–1815. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Niu, X.; Liang, F.; Dai, Y.; Liang, J.; Li, J.; Wu, X.; Zheng, H.; Qi, T.; Sheng, W. Rnf213 loss-of-function promotes pathological angiogenesis in moyamoya disease via the hippo pathway. Brain 2023, 146, 4674–4689. [Google Scholar] [CrossRef] [PubMed]
- Schaheen, B.; Downs, E.A.; Serbulea, V.; Almenara, C.C.; Spinosa, M.; Su, G.; Zhao, Y.; Srikakulapu, P.; Butts, C.; McNamara, C.A.; et al. B-cell depletion promotes aortic infiltration of immunosuppressive cells and is protective of experimental aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, C.; Cecchi, I.; Barbarroja, N.; Patino-Trives, A.M.; Luque-Tevar, M.; Perez-Sanchez, L.; Ibáñez-Costa, A.; de la Rosa, I.A.; Ortega, R.; Escudero, A.; et al. Early restoration of immune and vascular phenotypes in systemic lupus erythematosus and rheumatoid arthritis patients after b cell depletion. J. Cell Mol. Med. 2019, 23, 6308–6318. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Chan, J.K.Y. Endothelial function and endothelial progenitor cells in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2022, 18, 286–300. [Google Scholar] [CrossRef]
- Corban, M.T.; Duarte-Garcia, A.; McBane, R.D.; Matteson, E.L.; Lerman, L.O.; Lerman, A. Antiphospholipid syndrome: Role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. J. Am. Coll. Cardiol. 2017, 69, 2317–2330. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, J.H.; Phi, J.H.; Kim, Y.Y.; Kim, J.E.; Wang, K.C.; Cho, B.K.; Kim, S.K. Plasma matrix metalloproteinases, cytokines and angiogenic factors in moyamoya disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 673–678. [Google Scholar] [CrossRef]
- Liu, W.; Sun, J.; Shi, Z.; Huang, Z.; Yu, L.; Du, H.; Ge, P.; Zhang, D. Circulating inflammatory cytokine associated with poor prognosis in moyamoya disease: A prospective cohort study. J. Clin. Med. 2023, 12, 823. [Google Scholar] [CrossRef]
- Lee, C.Y.; Wu, S.W.; Yang, J.J.; Chen, W.Y.; Chen, C.J.; Chen, H.H.; Lee, Y.C.; Su, C.H.; Kuan, Y.H. Vascular endothelial dysfunction induced by 3-bromofluoranthene via mapk-mediated-nfkappab pro-inflammatory pathway and intracellular ros generation. Arch. Toxicol. 2024, 98, 2247–2259. [Google Scholar] [CrossRef]
- Dong, X.; Wu, D.; Zhang, Y.; Jia, L.; Pan, X.; Sun, J.; Pan, L.L. Cathelicidin modulates vascular smooth muscle cell phenotypic switching through ros/il-6 pathway. Antioxidants 2020, 9, 491. [Google Scholar] [CrossRef]
- Chen, T.; Wei, W.; Zhang, J.; Yu, J.; Xu, S.; Wu, D.; Li, X.; Chen, J. Assessment of plasma soluble tie-2 level to distinguish moyamoya disease from atherosclerotic cerebrovascular disease and predict postoperative neovascularization. J. Neurosurg. 2023, 139, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The role of the vegf family in atherosclerosis development and its potential as treatment targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. Il-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating hif-1alpha and vegf expression in vascular endothelial cells. EBioMedicine 2018, 33, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Chung, J.W.; Kim, D.H.; Won, H.H.; Yeon, J.Y.; Ki, C.S.; Shin, H.J.; Kim, J.S.; Hong, S.C.; Kim, D.K.; et al. Moyamoya disease and spectrums of rnf213 vasculopathy. Transl. Stroke Res. 2020, 11, 580–589. [Google Scholar] [CrossRef]
- Hiraide, T.; Suzuki, H.; Momoi, M.; Shinya, Y.; Fukuda, K.; Kosaki, K.; Kataoka, M. Rnf213-associated vascular disease: A concept unifying various vasculopathies. Life 2022, 12, 555. [Google Scholar] [CrossRef]
- Roy, V.; Ross, J.P.; Pepin, R.; Ghio, S.C.; Brodeur, A.; Deschenes, L.T.; Le-Bel, G.; Phillips, D.E.; Milot, G.; Dion, P.A.; et al. Moyamoya disease susceptibility gene rnf213 regulates endothelial barrier function. Stroke 2022, 53, 1263–1275. [Google Scholar] [CrossRef]
- Chiablaem, K.; Jinawath, A.; Nuanpirom, J.; Arora, J.K.; Nasaree, S.; Thanomchard, T.; Singhto, N.; Chittavanich, P.; Suktitipat, B.; Charoensawan, V.; et al. Identification of rnf213 as a potential suppressor of local invasion in intrahepatic cholangiocarcinoma. Lab. Investig. 2024, 104, 102074. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Jiang, Q.; Liu, Y.; Zhang, X.; Huang, Y.; Zhang, H. The Role of Immune Cells in Moyamoya Disease. Brain Sci. 2025, 15, 137. https://doi.org/10.3390/brainsci15020137
Wang S, Jiang Q, Liu Y, Zhang X, Huang Y, Zhang H. The Role of Immune Cells in Moyamoya Disease. Brain Sciences. 2025; 15(2):137. https://doi.org/10.3390/brainsci15020137
Chicago/Turabian StyleWang, Sheng, Qian Jiang, Yuan Liu, Xincheng Zhang, Yimin Huang, and Huaqiu Zhang. 2025. "The Role of Immune Cells in Moyamoya Disease" Brain Sciences 15, no. 2: 137. https://doi.org/10.3390/brainsci15020137
APA StyleWang, S., Jiang, Q., Liu, Y., Zhang, X., Huang, Y., & Zhang, H. (2025). The Role of Immune Cells in Moyamoya Disease. Brain Sciences, 15(2), 137. https://doi.org/10.3390/brainsci15020137