
Models of Trigeminal Activation: Is There an Animal Model of Migraine?



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Multifaceted World of Migraine: Characteristics, Pathomechanisms, Associated Disorders, and Therapeutic Challenges
3. Exploring Animal Models in Migraine Research: Unraveling Complexity
3.1. Cranial Stimulation Models: Investigating Migraine Mechanisms via Electrical Activation
3.2. Migraine Models: Chemical Stimulation of the Dura Mater
3.3. Exploring the Migraine-Inducing Properties of Nitroglycerin: Mechanisms, Neurological Impacts, and Experimental Models
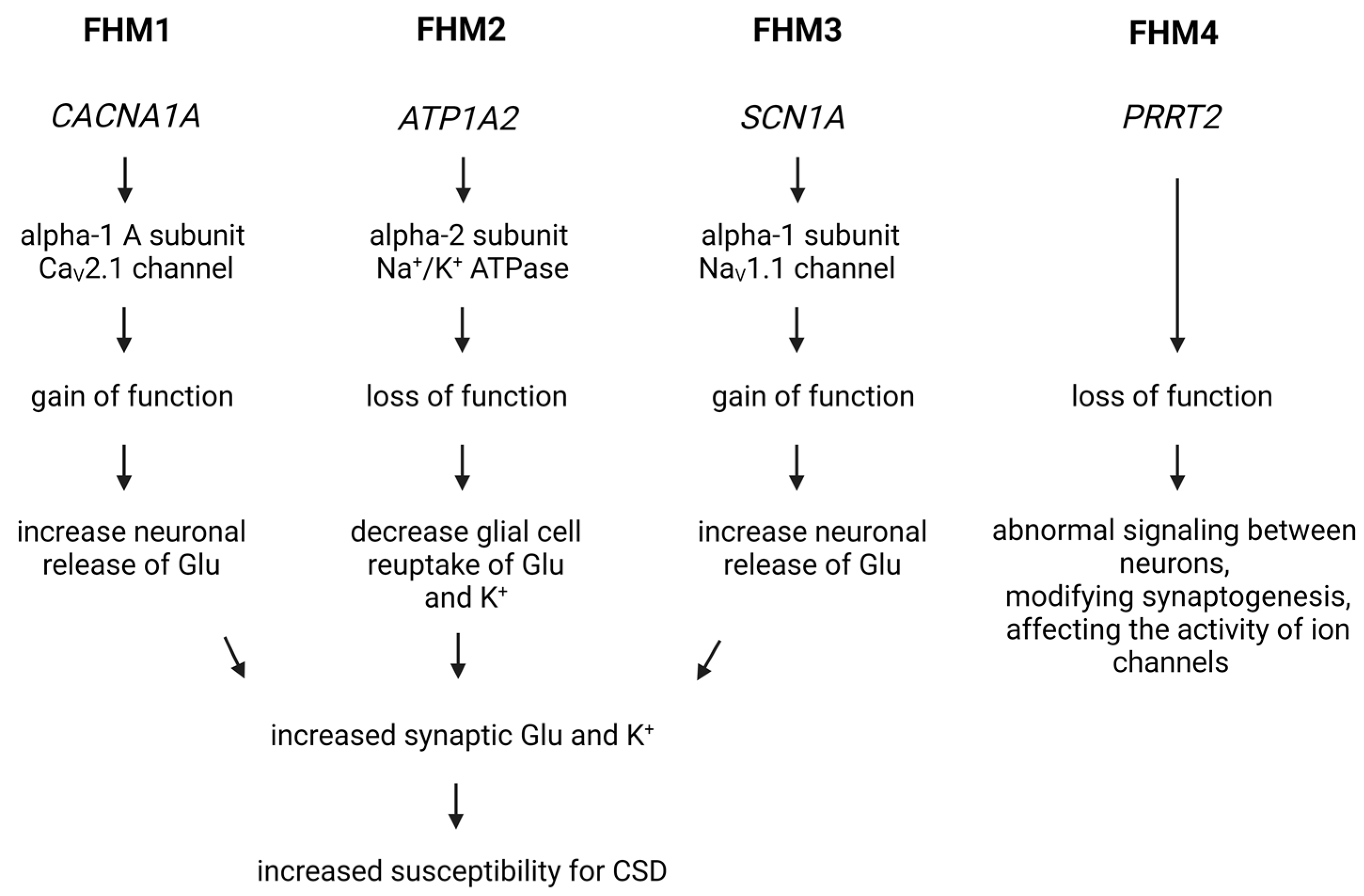
3.4. The Role of Genetic Factors in the Development of Migraine: Models and Research Approaches
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT1B/D receptor | 5-hydroxytryptamine 1B/1D receptor |
| CAMKIIα | Calcium/calmodulin-dependent protein kinase II alfa |
| CFA | Complete Freund’s adjuvant |
| cGMP | Cyclic guanosine monophosphate |
| CGRP | Calcitonin gene-related peptide |
| CM | Chronic migraine |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CSD | Cortical spreading depression |
| EM | Episodic migraine |
| eNOS | Endothelial nitric oxide synthase |
| FHM | Familial hemiplegic migraine |
| GWAS | Genome-wide association studies |
| ICHD-3 | The International Classification of Headache Disorders-3 beta |
| iNOS | Inducible nitric oxide synthase |
| IS | Inflammatory soup |
| KP | Kynurenine pathway |
| MOH | Medication-overuse headache |
| NF-κB | Nuclear factor kappa B |
| NI | Neurogenic inflammation |
| NMDA | N-methyl-D-aspartate |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| NTG | Nitroglycerin |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| PRS | Polygenic risk score |
| sGC | enzyme-soluble guanylyl cyclase |
| SSS | Superior sagittal sinus |
| TNC | Caudal trigeminal nucleus |
| TRPV1 | Transient receptor potential vanilloid type 1 |
| VIP | Vasoactive intestinal peptide |
References
- Leonardi, M.; Steiner, T.J.; Scher, A.T.; Lipton, R.B. The global burden of migraine: Measuring disability in headache disorders with WHO’s Classification of Functioning, Disability and Health (ICF). J. Headache Pain 2005, 6, 429–440. [Google Scholar] [CrossRef]
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, S.; Adjepong, D.; Al-Shami, H.A.; Malik, B.H. Is There an Association between Migraine and Major Depressive Disorder? A Narrative Review. Cureus 2020, 12, e8551. [Google Scholar] [CrossRef] [PubMed]
- Al-Khazali, H.M.; Younis, S.; Al-Sayegh, Z.; Ashina, S.; Ashina, M.; Schytz, H.W. Prevalence of neck pain in migraine: A systematic review and meta-analysis. Cephalalgia 2022, 42, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Kelman, L. The premonitory symptoms (prodrome): A tertiary care study of 893 migraineurs. Headache 2004, 44, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Schoonman, G.G.; Evers, D.J.; Terwindt, G.M.; van Dijk, J.G.; Ferrari, M.D. The prevalence of premonitory symptoms in migraine: A questionnaire study in 461 patients. Cephalalgia 2006, 26, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Lipton, R.B.; Dodick, D.W.; Silberstein, S.D.; Saper, J.R.; Aurora, S.K.; Goadsby, P.J.; Charles, A. Migraine headache is present in the aura phase: A prospective study. Neurology 2012, 79, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Maniyar, F.H.; Sprenger, T.; Monteith, T.; Schankin, C.; Goadsby, P.J. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain 2014, 137 Pt 1, 232–241. [Google Scholar] [CrossRef]
- Burstein, R.; Noseda, R.; Borsook, D. Migraine: Multiple processes, complex pathophysiology. J. Neurosci. 2015, 35, 6619–6629. [Google Scholar] [CrossRef]
- Khan, J.; Asoom, L.I.A.; Sunni, A.A.; Rafique, N.; Latif, R.; Saif, S.A.; Almandil, N.B.; Almohazey, D.; AbdulAzeez, S.; Borgio, J.F. Genetics, pathophysiology, diagnosis, treatment, management, and prevention of migraine. Biomed. Pharmacother. 2021, 139, 111557. [Google Scholar] [CrossRef]
- Kelman, L.; Tanis, D. The relationship between migraine pain and other associated symptoms. Cephalalgia 2006, 26, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Bahra, A. Primary Headache Disorders: Focus on Migraine. Rev. Pain 2011, 5, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Katsarava, Z.; Buse, D.C.; Manack, A.N.; Lipton, R.B. Defining the differences between episodic migraine and chronic migraine. Curr. Pain Headache Rep. 2012, 16, 86–92. [Google Scholar] [CrossRef]
- Lipton, R.B. Tracing transformation: Chronic migraine classification, progression, and epidemiology. Neurology 2009, 72 (Suppl. S5), S3–S7. [Google Scholar] [CrossRef]
- Ashina, S.; Serrano, D.; Lipton, R.B.; Maizels, M.; Manack, A.N.; Turkel, C.C.; Reed, M.L.; Buse, D.C. Depression and risk of transformation of episodic to chronic migraine. J. Headache Pain 2012, 13, 615–624. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L. Tracing neural connections to pain pathways with relevance to primary headaches. Cephalalgia 2011, 31, 737–747. [Google Scholar] [CrossRef]
- Fusco, B.M.; Barzoi, G.; Agrò, F. Repeated intranasal capsaicin applications to treat chronic migraine. Br. J. Anaesth. 2003, 90, 812. [Google Scholar] [CrossRef]
- Matharu, M.S.; Bartsch, T.; Ward, N.; Frackowiak, R.S.; Weiner, R.; Goadsby, P.J. Central neuromodulation in chronic migraine patients with suboccipital stimulators: A PET study. Brain 2004, 127, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Hadjikhani, N. The cerebellum and migraine. Headache 2007, 47, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Spekker, E.; Nagy-Grócz, G.; Vécsei, L. Ion Channel Disturbances in Migraine Headache: Exploring the Potential Role of the Kynurenine System in the Context of the Trigeminovascular System. Int. J. Mol. Sci. 2023, 24, 16574. [Google Scholar] [CrossRef]
- Weiller, C.; May, A.; Limmroth, V.; Jüptner, M.; Kaube, H.; Schayck, R.V.; Coenen, H.H.; Diener, H.C. Brain stem activation in spontaneous human migraine attacks. Nat. Med. 1995, 1, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Jakubowski, M.; Garcia-Nicas, E.; Kainz, V.; Bajwa, Z.; Hargreaves, R.; Becerra, L.; Borsook, D. Thalamic sensitization transforms localized pain into widespread allodynia. Ann. Neurol. 2010, 68, 81–91. [Google Scholar] [CrossRef]
- Moulton, E.A.; Becerra, L.; Maleki, N.; Pendse, G.; Tully, S.; Hargreaves, R.; Burstein, R.; Borsook, D. Painful heat reveals hyperexcitability of the temporal pole in interictal and ictal migraine States. Cereb. Cortex 2011, 21, 435–448. [Google Scholar] [CrossRef]
- Maleki, N.; Becerra, L.; Nutile, L.; Pendse, G.; Brawn, J.; Bigal, M.; Burstein, R.; Borsook, D. Migraine attacks the Basal Ganglia. Mol. Pain 2011, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Russo, A.; Esposito, F.; Giordano, A.; Taglialatela, G.; De Micco, R.; Cirillo, M.; Conte, F.; d’Onofrio, F.; Cirillo, S.; et al. Interictal cortical reorganization in episodic migraine without aura: An event-related fMRI study during parametric trigeminal nociceptive stimulation. Neurol. Sci. 2011, 32 (Suppl. S1), S165–S167. [Google Scholar] [CrossRef]
- DaSilva, A.F.; Granziera, C.; Snyder, J.; Hadjikhani, N. Thickening in the somatosensory cortex of patients with migraine. Neurology 2007, 69, 1990–1995. [Google Scholar] [CrossRef]
- Hadjikhani, N. Relevance of cortical thickness in migraine sufferers. Expert. Rev. Neurother. 2008, 8, 327–329. [Google Scholar] [CrossRef]
- Valfrè, W.; Rainero, I.; Bergui, M.; Pinessi, L. Voxel-based morphometry reveals gray matter abnormalities in migraine. Headache 2008, 48, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Raecke, R.; Niemeier, A.; Ihle, K.; Ruether, W.; May, A. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J. Neurosci. 2009, 29, 13746–13750. [Google Scholar] [CrossRef] [PubMed]
- Baliki, M.N.; Schnitzer, T.J.; Bauer, W.R.; Apkarian, A.V. Brain morphological signatures for chronic pain. PLoS ONE 2011, 6, e26010. [Google Scholar] [CrossRef]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.G.; Tournier-Lasserve, E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef] [PubMed]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; van den Maagdenberg, A.M.; Pusch, M.; et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 366, 371–377. [Google Scholar] [CrossRef]
- The International Headache Genetics Consortium. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 2010, 42, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Schürks, M.; Anttila, V.; de Vries, B.; Schminke, U.; Launer, L.J.; Terwindt, G.M.; van den Maagdenberg, A.M.; Fendrich, K.; Völzke, H.; et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 2011, 43, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Freilinger, T.; Anttila, V.; de Vries, B.; Malik, R.; Kallela, M.; Terwindt, G.M.; Pozo-Rosich, P.; Winsvold, B.; Nyholt, D.R.; van Oosterhout, W.P.; et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012, 44, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Albury, C.L.; Griffiths, L.R. Advances in genetics of migraine. J. Headache Pain 2019, 20, 72. [Google Scholar] [CrossRef]
- Peroutka, S.J. What turns on a migraine? A systematic review of migraine precipitating factors. Curr. Pain Headache Rep. 2014, 18, 454. [Google Scholar] [CrossRef]
- Pavlovic, J.M.; Buse, D.C.; Sollars, C.M.; Haut, S.; Lipton, R.B. Trigger factors and premonitory features of migraine attacks: Summary of studies. Headache 2014, 54, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Marmura, M.J. Triggers, Protectors, and Predictors in Episodic Migraine. Curr. Pain Headache Rep. 2018, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- Vgontzas, A.; Pavlović, J.M. Sleep Disorders and Migraine: Review of Literature and Potential Pathophysiology Mechanisms. Headache 2018, 58, 1030–1039. [Google Scholar] [CrossRef]
- Martinelli, D.; Pocora, M.M.; De Icco, R.; Putortì, A.; Tassorelli, C. Triggers of migraine: Where do we stand? Curr. Opin. Neurol. 2022, 35, 360–366. [Google Scholar] [CrossRef]
- Buzzi, M.G.; Moskowitz, M.A. The trigemino-vascular system and migraine. Pathol. Biol. 1992, 40, 313–317. [Google Scholar]
- Buzzi, M.G.; Bonamini, M.; Moskowitz, M.A. Neurogenic model of migraine. Cephalalgia 1995, 15, 277–280. [Google Scholar] [CrossRef]
- Knyihár-Csillik, E.; Tajti, J.; Samsam, M.; Sáry, G.; Buzás, P.; Vécsei, L. Depletion of calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat after electrical stimulation of the Gasserian ganglion. Exp. Brain Res. 1998, 118, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Csillik, B.; Knyihár-Csillik, E.; Kreutzberg, G.W.; Tajti, L.; Kereszturi, A.; Kovács, T. Calcitonin gene-related peptide is released from cholinergic synapses. Ann. N. Y. Acad. Sci. 1992, 657, 466–468. [Google Scholar] [CrossRef]
- Csillik, B.; Tajti, L.; Kovács, T.; Kukla, E.; Rakic, P.; Knyihár-Csillik, E. Distribution of calcitonin gene-related peptide in vertebrate neuromuscular junctions: Relationship to the acetylcholine receptor. J. Histochem. Cytochem. 1993, 41, 1547–1555. [Google Scholar] [CrossRef]
- Bohár, Z.; Fejes-Szabó, A.; Tar, L.; Varga, H.; Tajti, J.; Párdutz, Á.; Vécsei, L. Evaluation of c-Fos immunoreactivity in the rat brainstem nuclei relevant in migraine pathogenesis after electrical stimulation of the trigeminal ganglion. Neurol. Sci. 2013, 34, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Körtési, T.; Tuka, B.; Tajti, J.; Bagoly, T.; Fülöp, F.; Helyes, Z.; Vécsei, L. Kynurenic Acid Inhibits the Electrical Stimulation Induced Elevated Pituitary Adenylate Cyclase-Activating Polypeptide Expression in the TNC. Front. Neurol. 2018, 8, 745. [Google Scholar] [CrossRef] [PubMed]
- Buzzi, M.G.; Carter, W.B.; Shimizu, T.; Heath, H.; Moskowitz, M.A. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology 1991, 30, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; De Icco, R.; Martinelli, D.; Putortì, A.; Tassorelli, C. Migraine neuroscience: From experimental models to target therapy. Neurol. Sci. 2020, 41 (Suppl. S2), 351–361. [Google Scholar] [CrossRef] [PubMed]
- Zagami, A.S.; Goadsby, P.J.; Edvinsson, L. Stimulation of the superior sagittal sinus in the cat causes release of vasoactive peptides. Neuropeptides 1990, 16, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.R.; Akerman, S.; Andreou, A.P.; Karsan, N.; Wemmie, J.A.; Goadsby, P.J. Acid-sensing ion channel 1: A novel therapeutic target for migraine with aura. Ann. Neurol. 2012, 72, 559–563. [Google Scholar] [CrossRef]
- Robert, C.; Bourgeais, L.; Arreto, C.D.; Condes-Lara, M.; Noseda, R.; Jay, T.; Villanueva, L. Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. J. Neurosci. 2013, 33, 8827–8840. [Google Scholar] [CrossRef] [PubMed]
- Kaube, H.; Keay, K.A.; Hoskin, K.L.; Bandler, R.; Goadsby, P.J. Expression of c-Fos-like immunoreactivity in the caudal medulla and upper cervical spinal cord following stimulation of the superior sagittal sinus in the cat. Brain Res. 1993, 629, 95–102. [Google Scholar] [CrossRef]
- Kaube, H.; Hoskin, K.L.; Goadsby, P.J. Acetylsalicylic acid inhibits cerebral cortical vasodilatation caused by superior sagittal sinus stimulation in the cat. Eur. J. Neurol. 1994, 1, 141–146. [Google Scholar] [CrossRef]
- Hoskin, K.L.; Goadsby, P.J. Comparison of more and less lipophilic serotonin (5HT1B/1D) agonists in a model of trigeminovascular nociception in cat. Exp. Neurol. 1998, 150, 45–51. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Hoskin, K.L. Differential effects of low dose CP122,288 and eletriptan on fos expression due to stimulation of the superior sagittal sinus in cat. Pain 1999, 82, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Strassman, A.; Mason, P.; Moskowitz, M.; Maciewicz, R. Response of brainstem trigeminal neurons to electrical stimulation of the dura. Brain Res. 1986, 379, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, M.; Messlinger, K.; Pawlak, M.; Schmidt, R.F. Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: Mediation by calcitonin gene-related peptide. Br. J. Pharmacol. 1995, 114, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Messlinger, K.; Hotta, H.; Pawlak, M.; Schmidt, R.F. Effects of the 5-HT1 receptor agonists, sumatriptan and CP 93,129, on dural arterial flow in the rat. Eur. J. Pharmacol. 1997, 332, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Phebus, L.A.; Johnson, K.W. Dural inflammation model of migraine pain. Curr. Protoc. Neurosci. 2001. [Google Scholar] [CrossRef]
- Burstein, R. Deconstructing migraine headache into peripheral and central sensitization. Pain 2001, 89, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lukács, M.; Haanes, K.A.; Majláth, Z.; Tajti, J.; Vécsei, L.; Warfvinge, K.; Edvinsson, L. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J. Headache Pain 2015, 16, 564. [Google Scholar] [CrossRef] [PubMed]
- Laborc, K.F.; Spekker, E.; Bohár, Z.; Szűcs, M.; Nagy-Grócz, G.; Fejes-Szabó, A.; Vécsei, L.; Párdutz, Á. Trigeminal activation patterns evoked by chemical stimulation of the dura mater in rats. J. Headache Pain 2020, 21, 101. [Google Scholar] [CrossRef] [PubMed]
- Spekker, E.; Tanaka, M.; Szabó, Á.; Vécsei, L. Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines 2021, 10, 76. [Google Scholar] [CrossRef]
- Cutrer, F.M.; Mitsikostas, D.D.; Ayata, G.; Sanchez del Rio, M. Attenuation by butalbital of capsaicin-induced c-fos-like immunoreactivity in trigeminal nucleus caudalis. Headache 1999, 39, 697–704. [Google Scholar] [CrossRef]
- Dux, M.; Sántha, P.; Jancsó, G. Capsaicin-sensitive neurogenic sensory vasodilatation in the dura mater of the rat. J. Physiol. 2003, 552 Pt 3, 859–867. [Google Scholar] [CrossRef]
- Jancsó, G. Intracisternal capsaicin: Selective degeneration of chemosensitive primary sensory afferents in the adult rat. Neurosci. Lett. 1981, 27, 41–45. [Google Scholar] [CrossRef]
- Szolcsányi, J. Antidromic vasodilatation and neurogenic inflammation. Agents Actions. 1988, 23, 4–11. [Google Scholar] [CrossRef]
- Kwon, Y.B.; Jeong, Y.C.; Kwon, J.K.; Son, J.S.; Kim, K.W. The Antinociceptive Effect of Sigma-1 Receptor Antagonist, BD1047, in a Capsaicin Induced Headache Model in Rats. Korean J. Physiol. Pharmacol. 2009, 13, 425–429. [Google Scholar] [CrossRef][Green Version]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K. Does inflammation have a role in migraine? Nat. Rev. Neurol. 2019, 15, 483–490. [Google Scholar] [CrossRef]
- Pardutz, A.; Krizbai, I.; Multon, S.; Vecsei, L.; Schoenen, J. Systemic nitroglycerin increases nNOS levels in rat trigeminal nucleus caudalis. Neuroreport 2000, 11, 3071–3075. [Google Scholar] [CrossRef]
- Geppetti, P.; Capone, J.G.; Trevisani, M.; Nicoletti, P.; Zagli, G.; Tola, M.R. CGRP and migraine: Neurogenic inflammation revisited. J. Headache Pain 2005, 6, 61–70. [Google Scholar] [CrossRef]
- Haanes, K.A.; Edvinsson, L. Pathophysiological Mechanisms in Migraine and the Identification of New Therapeutic Targets. CNS Drugs 2019, 33, 525–537. [Google Scholar] [CrossRef]
- Oshinsky, M.L.; Gomonchareonsiri, S. Episodic dural stimulation in awake rats: A model for recurrent headache. Headache 2007, 47, 1026–1036. [Google Scholar] [CrossRef]
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17. [Google Scholar] [CrossRef]
- Spekker, E.; Bohár, Z.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Estradiol Treatment Enhances Behavioral and Molecular Changes Induced by Repetitive Trigeminal Activation in a Rat Model of Migraine. Biomedicines 2022, 10, 3175. [Google Scholar] [CrossRef]
- Wieseler, J.; Ellis, A.; McFadden, A.; Stone, K.; Brown, K.; Cady, S.; Bastos, L.F.; Sprunger, D.; Rezvani, N.; Johnson, K.; et al. Supradural inflammatory soup in awake and freely moving rats induces facial allodynia that is blocked by putative immune modulators. Brain Res. 2017, 1664, 87–94. [Google Scholar] [CrossRef]
- Melo-Carrillo, A.; Lopez-Avila, A. A chronic animal model of migraine, induced by repeated meningeal nociception, characterized by a behavioral and pharmacological approach. Cephalalgia 2013, 33, 1096–1105. [Google Scholar] [CrossRef]
- Malick, A.; Jakubowski, M.; Elmquist, J.K.; Saper, C.B.; Burstein, R. A neurohistochemical blueprint for pain-induced loss of appetite. Proc. Natl. Acad. Sci. USA 2001, 98, 9930–9935. [Google Scholar] [CrossRef]
- Chen, N.; Su, W.; Cui, S.H.; Guo, J.; Duan, J.C.; Li, H.X.; He, L. A novel large animal model of recurrent migraine established by repeated administration of inflammatory soup into the dura mater of the rhesus monkey. Neural Regen. Res. 2019, 14, 100–106. [Google Scholar] [CrossRef]
- Parratt, J.R. Nitroglycerin-the first one hundred years: New facts about an old drug. J. Pharm. Pharmacol. 1979, 31, 801–809. [Google Scholar]
- Bektas, F.; Soyuncu, S. Nitroglycerin-induced migraine type headache: Bilaterally visible temporal arteries. Int. J. Emerg. Med. 2010, 3, 463–464. [Google Scholar] [CrossRef]
- Sicuteri, F.; Del Bene, E.; Poggioni, M.; Bonazzi, A. Unmasking latent dysnociception in healthy subjects. Headache 1987, 27, 180–185. [Google Scholar] [CrossRef]
- Sureda-Gibert, P.; Romero-Reyes, M.; Akerman, S. Nitroglycerin as a model of migraine: Clinical and preclinical review. Neurobiol. Pain 2022, 12, 100105. [Google Scholar] [CrossRef]
- Moncada, S. Adventures in vascular biology: A tale of two mediators. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 735–759. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Dominguez, J.M.; Muschamp, J.W.; Schmich, J.M.; Hull, E.M. Nitric oxide mediates glutamate-evoked dopamine release in the medial preoptic area. Neuroscience 2004, 125, 203–210. [Google Scholar] [CrossRef]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Kirkebøen, K.A.; Strand, O.A. The role of nitric oxide in sepsis—An overview. Acta Anaesthesiol. Scand. 1999, 43, 275–288. [Google Scholar] [CrossRef]
- Olesen, J.; Iversen, H.K.; Thomsen, L.L. Nitric oxide supersensitivity: A possible molecular mechanism of migraine pain. Neuroreport 1993, 4, 1027–1030. [Google Scholar] [CrossRef]
- Tassorelli, C.; Joseph, S.A.; Buzzi, M.G.; Nappi, G. The effects on the central nervous system of nitroglycerin—Putative mechanisms and mediators. Prog. Neurobiol. 1999, 57, 607–624. [Google Scholar] [CrossRef]
- Tassorelli, C.; Joseph, S.A.; Nappi, G. Reciprocal circuits involved in nitroglycerin-induced neuronal activation of autonomic regions and pain pathways: A double immunolabeling and tract-tracing study. Brain Res. 1999, 842, 294–310. [Google Scholar] [CrossRef]
- Iversen, H.K.; Olesen, J.; Tfelt-Hansen, P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain 1989, 38, 17–24. [Google Scholar] [CrossRef]
- Afridi, K.S.; Kaube, H.; Goadsby, J.P. Glyceryl trinitrate triggers premonitory symptoms in migraineurs. Pain 2004, 110, 675–680. [Google Scholar] [CrossRef]
- Sances, G.; Tassorelli, C.; Pucci, E.; Ghiotto, N.; Sandrini, G.; Nappi, G. Reliability of the nitroglycerin provocative test in the diagnosis of neurovascular headaches. Cephalalgia 2004, 24, 110–119. [Google Scholar] [CrossRef]
- Bahra, A.; Matharu, M.S.; Buchel, C.; Frackowiak, R.S.; Goadsby, P.J. Brainstem activation specific to migraine headache. Lancet 2001, 357, 1016–1017. [Google Scholar] [CrossRef]
- Juhasz, G.; Zsombok, T.; Modos, E.A.; Olajos, S.; Jakab, B.; Nemeth, J.; Szolcsanyi, J.; Vitrai, J.; Bagdy, G. NO-induced migraine attack: Strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain 2003, 106, 461–470. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann. Neurol. 1988, 23, 193–196. [Google Scholar] [CrossRef]
- Ashina, H.; Schytz, H.W.; Ashina, M. CGRP in human models of primary headaches. Cephalalgia 2018, 38, 353–360. [Google Scholar] [CrossRef]
- Juhasz, G.; Zsombok, T.; Jakab, B.; Nemeth, J.; Szolcsanyi, J.; Bagdy, G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia 2005, 25, 179–183. [Google Scholar] [CrossRef]
- Tassorelli, C.; Joseph, S.A.; Nappi, G. Central effects of nitroglycerin in the rat: New perspectives in migraine research. Funct. Neurol. 1996, 11, 219–223, 227–235. [Google Scholar]
- Pardutz, A.; Multon, S.; Malgrange, B.; Parducz, A.; Vecsei, L.; Schoenen, J. Effect of systemic nitroglycerin on CGRP and 5-HT afferents to rat caudal spinal trigeminal nucleus and its modulation by estrogen. Eur. J. Neurosci. 2002, 15, 1803–1809. [Google Scholar] [CrossRef]
- Pardutz, A.; Hoyk, Z.; Varga, H.; Vecsei, L.; Schoenen, J. Oestrogen-modulated increase of calmodulin-dependent protein kinase II (CamKII) in rat spinal trigeminal nucleus after systemic nitroglycerin. Cephalalgia 2007, 27, 46–53. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Tar, L.; Bohár, Z.; Fejes-Szabó, A.; Laborc, K.F.; Spekker, E.; Vécsei, L.; Párdutz, Á. The modulatory effect of anandamide on nitroglycerin-induced sensitization in the trigeminal system of the rat. Cephalalgia 2016, 36, 849–861. [Google Scholar] [CrossRef]
- Greco, R.; Tassorelli, C.; Cappelletti, D.; Sandrini, G.; Nappi, G. Activation of the transcription factor NF-kappaB in the nucleus trigeminalis caudalis in an animal model of migraine. Neurotoxicology 2005, 26, 795–800. [Google Scholar] [CrossRef]
- Varga, H.; Pardutz, A.; Vamos, E.; Bohar, Z.; Bago, F.; Tajti, J.; Bari, F.; Vecsei, L. Selective inhibition of cyclooxygenase-2 attenuates nitroglycerin-induced calmodulin-dependent protein kinase II alpha in rat trigeminal nucleus caudalis. Neurosci. Lett. 2009, 451, 170–173. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Bohár, Z.; Fejes-Szabó, A.; Laborc, K.F.; Spekker, E.; Tar, L.; Vécsei, L.; Párdutz, Á. Nitroglycerin increases serotonin transporter expression in rat spinal cord but anandamide modulated this effect. J. Chem. Neuroanat. 2017, 85, 13–20. [Google Scholar] [CrossRef]
- Fejes-Szabó, A.; Bohár, Z.; Vámos, E.; Nagy-Grócz, G.; Tar, L.; Veres, G.; Zádori, D.; Szentirmai, M.; Tajti, J.; Szatmári, I.; et al. Pre-treatment with new kynurenic acid amide dose-dependently prevents the nitroglycerine-induced neuronal activation and sensitization in cervical part of trigemino-cervical complex. J. Neural Transm. 2014, 121, 725–738. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur. J. Pharmacol. 1988, 154, 85–87. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2015, 17, 27. [Google Scholar] [CrossRef]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Laborc, K.F.; Veres, G.; Bajtai, A.; Bohár, Z.; Zádori, D.; Fejes-Szabó, A.; Spekker, E.; Vécsei, L.; Párdutz, Á. The Effect of Systemic Nitroglycerin Administration on the Kynurenine Pathway in the Rat. Front. Neurol. 2017, 8, 278, Erratum in Front. Neurol. 2020, 11, 1049. [Google Scholar] [CrossRef]
- Pradhan, A.A.; Smith, M.L.; McGuire, B.; Tarash, I.; Evans, C.J.; Charles, A. Characterization of a novel model of chronic migraine. Pain 2014, 155, 269–274. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Tassorelli, C. Chronic and intermittent administration of systemic nitroglycerin in the rat induces an increase in the gene expression of CGRP in central areas: Potential contribution to pain processing. J. Headache Pain 2018, 19, 51. [Google Scholar] [CrossRef]
- Tipton, A.F.; Tarash, I.; McGuire, B.; Charles, A.; Pradhan, A.A. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia 2016, 36, 1048–1056. [Google Scholar] [CrossRef]
- Sufka, K.J.; Staszko, S.M.; Johnson, A.P.; Davis, M.E.; Davis, R.E.; Smitherman, T.A. Clinically relevant behavioral endpoints in a recurrent nitroglycerin migraine model in rats. J. Headache Pain 2016, 17, 40. [Google Scholar] [CrossRef]
- Harris, H.M.; Carpenter, J.M.; Black, J.R.; Smitherman, T.A.; Sufka, K.J. The effects of repeated nitroglycerin administrations in rats; modeling migraine-related endpoints and chronification. J. Neurosci. Methods 2017, 284, 63–70. [Google Scholar] [CrossRef]
- Mahmoudi, J.; Mohaddes, G.; Erfani, M.; Sadigh-Eteghad, S.; Karimi, P.; Rajabi, M.; Reyhani-Rad, S.; Farajdokht, F. Cerebrolysin attenuates hyperalgesia, photophobia, and neuroinflammation in a nitroglycerin-induced migraine model in rats. Brain Res. Bull. 2018, 140, 197–204. [Google Scholar] [CrossRef]
- Tuka, B.; Helyes, Z.; Markovics, A.; Bagoly, T.; Szolcsányi, J.; Szabó, N.; Tóth, E.; Kincses, Z.T.; Vécsei, L.; Tajti, J. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 2013, 33, 1085–1095. [Google Scholar] [CrossRef]
- Russell, M.B.; Hilden, J.; Sørensen, S.A.; Olesen, J. Familial occurrence of migraine without aura and migraine with aura. Neurology 1993, 43, 1369–1373. [Google Scholar] [CrossRef]
- Grangeon, L.; Lange, K.S.; Waliszewska-Prosół, M.; Onan, D.; Marschollek, K.; Wiels, W.; Mikulenka, P.; Farham, F.; Gollion, C.; on behalf of the European Headache Federation School of Advanced Studies (EHF-SAS). Genetics of migraine: Where are we now? J. Headache Pain 2023, 24, 12. [Google Scholar] [CrossRef]
- Dehghani, A.; Karatas, H. Mouse Models of Familial Hemiplegic Migraine for Studying Migraine Pathophysiology. Curr. Neuropharmacol. 2019, 17, 961–973. [Google Scholar] [CrossRef]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998, 24, 307–323. [Google Scholar] [CrossRef]
- Tottene, A.; Fellin, T.; Pagnutti, S.; Luvisetto, S.; Striessnig, J.; Fletcher, C.; Pietrobon, D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 13284–13289. [Google Scholar] [CrossRef]
- van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Adams, P.J.; Rungta, R.L.; Garcia, E.; van den Maagdenberg, A.M.; MacVicar, B.A.; Snutch, T.P. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 18694–18699. [Google Scholar] [CrossRef]
- Stam, A.H.; Luijckx, G.J.; Poll-Thé, B.T.; Ginjaar, I.B.; Frants, R.R.; Haan, J.; Ferrari, M.D.; Terwindt, G.M.; van den Maagdenberg, A.M. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1125–1129. [Google Scholar] [CrossRef]
- Eikermann-Haerter, K.; Dileköz, E.; Kudo, C.; Savitz, S.I.; Waeber, C.; Baum, M.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; Moskowitz, M.A.; Ayata, C. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Investig. 2009, 119, 99–109. [Google Scholar] [CrossRef]
- Langford, D.J.; Bailey, A.L.; Chanda, M.L.; Clarke, S.E.; Drummond, T.E.; Echols, S.; Glick, S.; Ingrao, J.; Klassen-Ross, T.; Lacroix-Fralish, M.L.; et al. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 2010, 7, 447–449. [Google Scholar] [CrossRef]
- Chanda, M.L.; Tuttle, A.H.; Baran, I.; Atlin, C.; Guindi, D.; Hathaway, G.; Israelian, N.; Levenstadt, J.; Low, D.; Macrae, L.; et al. Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain 2013, 154, 1254–1262. [Google Scholar] [CrossRef]
- Fioretti, B.; Catacuzzeno, L.; Sforna, L.; Gerke-Duncan, M.B.; van den Maagdenberg, A.M.; Franciolini, F.; Connor, M.; Pietrobon, D. Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 2011, 589 Pt 23, 5879–5895. [Google Scholar] [CrossRef]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; van den Maagdenberg, A.M.; Ferrari, M.D.; Pietrobon, D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef]
- Vecchia, D.; Tottene, A.; van den Maagdenberg, A.M.; Pietrobon, D. Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol. Dis. 2014, 69, 225–234. [Google Scholar] [CrossRef]
- Vecchia, D.; Tottene, A.; van den Maagdenberg, A.M.; Pietrobon, D. Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front. Cell Neurosci. 2015, 9, 8. [Google Scholar] [CrossRef]
- Loonen, I.C.M.; Voskuyl, R.A.; Schenke, M.; van Heiningen, S.H.; van den Maagdenberg, A.M.J.M.; Tolner, E.A. Spontaneous and optogenetically induced cortical spreading depolarization in familial hemiplegic migraine type 1 mutant mice. Neurobiol. Dis. 2024, 192, 106405. [Google Scholar] [CrossRef]
- Klychnikov, O.I.; Li, K.W.; Sidorov, I.A.; Loos, M.; Spijker, S.; Broos, L.A.; Frants, R.R.; Ferrari, M.D.; Mayboroda, O.A.; Deelder, A.M.; et al. Quantitative cortical synapse proteomics of a transgenic migraine mouse model with mutated Ca(v)2.1 calcium channels. Proteomics 2010, 10, 2531–2535. [Google Scholar] [CrossRef]
- Di Guilmi, M.N.; Wang, T.; Inchauspe, C.G.; Forsythe, I.D.; Ferrari, M.D.; van den Maagdenberg, A.M.; Borst, J.G.; Uchitel, O.D. Synaptic gain-of-function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J. Neurosci. 2014, 34, 7047–7058. [Google Scholar] [CrossRef]
- Inchauspe, C.G.; Urbano, F.J.; Di Guilmi, M.N.; Ferrari, M.D.; van den Maagdenberg, A.M.; Forsythe, I.D.; Uchitel, O.D. Presynaptic CaV2.1 calcium channels carrying familial hemiplegic migraine mutation R192Q allow faster recovery from synaptic depression in mouse calyx of Held. J. Neurophysiol. 2012, 108, 2967–2976. [Google Scholar] [CrossRef][Green Version]
- Dilekoz, E.; Houben, T.; Eikermann-Haerter, K.; Balkaya, M.; Lenselink, A.M.; Whalen, M.J.; Spijker, S.; Ferrari, M.D.; van den Maagdenberg, A.M.; Ayata, C. Migraine mutations impair hippocampal learning despite enhanced long-term potentiation. J. Neurosci. 2015, 35, 3397–3402. [Google Scholar] [CrossRef][Green Version]
- Hullugundi, S.K.; Ansuini, A.; Ferrari, M.D.; van den Maagdenberg, A.M.; Nistri, A. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience 2014, 266, 244–254. [Google Scholar] [CrossRef]
- Franceschini, A.; Vilotti, S.; Ferrari, M.D.; van den Maagdenberg, A.M.; Nistri, A.; Fabbretti, E. TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS ONE 2013, 8, e52394. [Google Scholar] [CrossRef]
- Eising, E.; Shyti, R.; ‘t Hoen, P.A.C.; Vijfhuizen, L.S.; Huisman, S.M.H.; Broos, L.A.M.; Mahfouz, A.; Reinders, M.J.T.; Ferrari, M.D.; Tolner, E.A.; et al. Cortical Spreading Depression Causes Unique Dysregulation of Inflammatory Pathways in a Transgenic Mouse Model of Migraine. Mol. Neurobiol. 2017, 54, 2986–2996. [Google Scholar] [CrossRef]
- Shyti, R.; Eikermann-Haerter, K.; van Heiningen, S.H.; Meijer, O.C.; Ayata, C.; Joëls, M.; Ferrari, M.D.; van den Maagdenberg, A.M.; Tolner, E.A. Stress hormone corticosterone enhances susceptibility to cortical spreading depression in familial hemiplegic migraine type 1 mutant mice. Exp. Neurol. 2015, 263, 214–220. [Google Scholar] [CrossRef]
- Carreira, R.J.; Shyti, R.; Balluff, B.; Abdelmoula, W.M.; van Heiningen, S.H.; van Zeijl, R.J.; Dijkstra, J.; Ferrari, M.D.; Tolner, E.A.; McDonnell, L.A.; et al. Large-scale mass spectrometry imaging investigation of consequences of cortical spreading depression in a transgenic mouse model of migraine. J. Am. Soc. Mass. Spectrom. 2015, 26, 853–861. [Google Scholar] [CrossRef][Green Version]
- Shyti, R.; Kohler, I.; Schoenmaker, B.; Derks, R.J.; Ferrari, M.D.; Tolner, E.A.; Mayboroda, O.A.; van den Maagdenberg, A.M. Plasma metabolic profiling after cortical spreading depression in a transgenic mouse model of hemiplegic migraine by capillary electrophoresis–mass spectrometry. Mol. Biosyst. 2015, 11, 1462–1471. [Google Scholar] [CrossRef]
- Deboer, T.; van Diepen, H.C.; Ferrari, M.D.; van den Maagdenberg, A.M.; Meijer, J.H. Reduced sleep and low adenosinergic sensitivity in cacna1a R192Q mutant mice. Sleep 2013, 36, 127–136. [Google Scholar] [CrossRef]
- Eikermann-Haerter, K.; Yuzawa, I.; Dilekoz, E.; Joutel, A.; Moskowitz, M.A.; Ayata, C. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy syndrome mutations increase susceptibility to spreading depression. Ann. Neurol. 2011, 69, 413–418. [Google Scholar] [CrossRef]
- Eikermann-Haerter, K.; Baum, M.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; Moskowitz, M.A.; Ayata, C. Androgenic suppression of spreading depression in familial hemiplegic migraine type 1 mutant mice. Ann. Neurol. 2009, 66, 564–568. [Google Scholar] [CrossRef]
- Tavraz, N.N.; Friedrich, T.; Dürr, K.L.; Koenderink, J.B.; Bamberg, E.; Freilinger, T.; Dichgans, M. Diverse functional consequences of mutations in the Na+/K+-ATPase α2-subunit causing familial hemiplegic migraine type 2. J. Biol. Chem. 2008, 283, 31097–31106. [Google Scholar] [CrossRef]
- Pisano, T.; Spiller, S.; Mei, D.; Guerrini, R.; Cianchetti, C.; Friedrich, T.; Pruna, D. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia 2013, 33, 1302–1310. [Google Scholar] [CrossRef]
- Leo, L.; Gherardini, L.; Barone, V.; De Fusco, M.; Pietrobon, D.; Pizzorusso, T.; Casari, G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2011, 7, e1002129. [Google Scholar] [CrossRef]
- Bøttger, P.; Glerup, S.; Gesslein, B.; Illarionova, N.B.; Isaksen, T.J.; Heuck, A.; Clausen, B.H.; Füchtbauer, E.M.; Gramsbergen, J.B.; Gunnarson, E.; et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016, 6, 22047. [Google Scholar] [CrossRef]
- Unekawa, M.; Ikeda, K.; Tomita, Y.; Kawakami, K.; Suzuki, N. Enhanced susceptibility to cortical spreading depression in two types of Na+,K+-ATPase α2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 2018, 38, 1515–1524. [Google Scholar] [CrossRef]
- Smith, S.E.; Chen, X.; Brier, L.M.; Bumstead, J.R.; Rensing, N.R.; Ringel, A.E.; Shin, H.; Oldenborg, A.; Crowley, J.R.; Bice, A.R.; et al. Astrocyte deletion of α2-Na/K ATPase triggers episodic motor paralysis in mice via a metabolic pathway. Nat. Commun. 2020, 11, 6164. [Google Scholar] [CrossRef]
- Capuani, C.; Melone, M.; Tottene, A.; Bragina, L.; Crivellaro, G.; Santello, M.; Casari, G.; Conti, F.; Pietrobon, D. Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 2016, 8, 967–986. [Google Scholar] [CrossRef]
- Martin, M.S.; Dutt, K.; Papale, L.A.; Dubé, C.M.; Dutton, S.B.; de Haan, G.; Shankar, A.; Tufik, S.; Meisler, M.H.; Baram, T.Z.; et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 2010, 285, 9823–9834. [Google Scholar] [CrossRef]
- Cestèle, S.; Scalmani, P.; Rusconi, R.; Terragni, B.; Franceschetti, S.; Mantegazza, M. Self-limited hyperexcitability: Functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J. Neurosci. 2008, 28, 7273–7283. [Google Scholar] [CrossRef]
- Bertelli, S.; Barbieri, R.; Pusch, M.; Gavazzo, P. Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA. Cephalalgia 2019, 39, 477–488. [Google Scholar] [CrossRef]
- Jansen, N.A.; Dehghani, A.; Linssen, M.M.L.; Breukel, C.; Tolner, E.A.; van den Maagdenberg, A.M.J.M. First FHM3 mouse model shows spontaneous cortical spreading depolarizations. Ann. Clin. Transl. Neurol. 2020, 7, 132–138. [Google Scholar] [CrossRef]
- Auffenberg, E.; Hedrich, U.B.; Barbieri, R.; Miely, D.; Groschup, B.; Wuttke, T.V.; Vogel, N.; Lührs, P.; Zanardi, I.; Bertelli, S.; et al. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J. Clin. Investig. 2021, 131, e142202. [Google Scholar] [CrossRef]
- Michetti, C.; Castroflorio, E.; Marchionni, I.; Forte, N.; Sterlini, B.; Binda, F.; Fruscione, F.; Baldelli, P.; Valtorta, F.; Zara, F.; et al. The PRRT2 knockout mouse recapitulates the neurological diseases associated with PRRT2 mutations. Neurobiol. Dis. 2017, 99, 66–83. [Google Scholar] [CrossRef]
- Valente, P.; Romei, A.; Fadda, M.; Sterlini, B.; Lonardoni, D.; Forte, N.; Fruscione, F.; Castroflorio, E.; Michetti, C.; Giansante, G.; et al. Constitutive Inactivation of the PRRT2 Gene Alters Short-Term Synaptic Plasticity and Promotes Network Hyperexcitability in Hippocampal Neurons. Cereb. Cortex 2019, 29, 2010–2033. [Google Scholar] [CrossRef]
- Savino, E.; Guarnieri, F.C.; Tsai, J.W.; Corradi, A.; Benfenati, F.; Valtorta, F. An Emerging Role of PRRT2 in Regulating Growth Cone Morphology. Cells 2021, 10, 2666. [Google Scholar] [CrossRef]
- Lafrenière, R.G.; Cader, M.Z.; Poulin, J.F.; Andres-Enguix, I.; Simoneau, M.; Gupta, N.; Boisvert, K.; Lafrenière, F.; McLaughlan, S.; Dubé, M.P.; et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 2010, 16, 1157–1160. [Google Scholar] [CrossRef]
- Brennan, K.C.; Bates, E.A.; Shapiro, R.E.; Zyuzin, J.; Hallows, W.C.; Huang, Y.; Lee, H.Y.; Jones, C.R.; Fu, Y.H.; Charles, A.C.; et al. Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci. Transl. Med. 2013, 5, 183ra56. [Google Scholar] [CrossRef]
- Tan, R.Y.; Markus, H.S. CADASIL: Migraine, Encephalopathy, Stroke and Their Inter-Relationships. PLoS ONE 2016, 11, e0157613. [Google Scholar] [CrossRef]
- Weir, G.A.; Pettingill, P.; Wu, Y.; Duggal, G.; Ilie, A.S.; Akerman, C.J.; Cader, M.Z. The Role of TRESK in Discrete Sensory Neuron Populations and Somatosensory Processing. Front. Mol. Neurosci. 2019, 12, 170. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spekker, E.; Fejes-Szabó, A.; Nagy-Grócz, G. Models of Trigeminal Activation: Is There an Animal Model of Migraine? Brain Sci. 2024, 14, 317. https://doi.org/10.3390/brainsci14040317
Spekker E, Fejes-Szabó A, Nagy-Grócz G. Models of Trigeminal Activation: Is There an Animal Model of Migraine? Brain Sciences. 2024; 14(4):317. https://doi.org/10.3390/brainsci14040317
Chicago/Turabian StyleSpekker, Eleonóra, Annamária Fejes-Szabó, and Gábor Nagy-Grócz. 2024. "Models of Trigeminal Activation: Is There an Animal Model of Migraine?" Brain Sciences 14, no. 4: 317. https://doi.org/10.3390/brainsci14040317
APA StyleSpekker, E., Fejes-Szabó, A., & Nagy-Grócz, G. (2024). Models of Trigeminal Activation: Is There an Animal Model of Migraine? Brain Sciences, 14(4), 317. https://doi.org/10.3390/brainsci14040317