

The Comprehensive Analysis of m6A-Associated Anoikis Genes in Low-Grade Gliomas
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of Data
2.2. Differential Expression Analysis and Functional Enrichment Analysis
2.3. Analysis of Protein-Protein Interaction (PPI)
2.4. Construction of the Risk Model
2.5. Analyses of Risk Model Genes
2.6. Clinical Correlation Analysis
2.7. Construction of the Independent Prognostic Model
2.8. Immune Infiltration Analysis and Drug Susceptibility Analysis
2.9. Construction of Competing Endogenous RNA (ceRNA) Network
2.10. External Validation of Risk Model Genes
3. Results
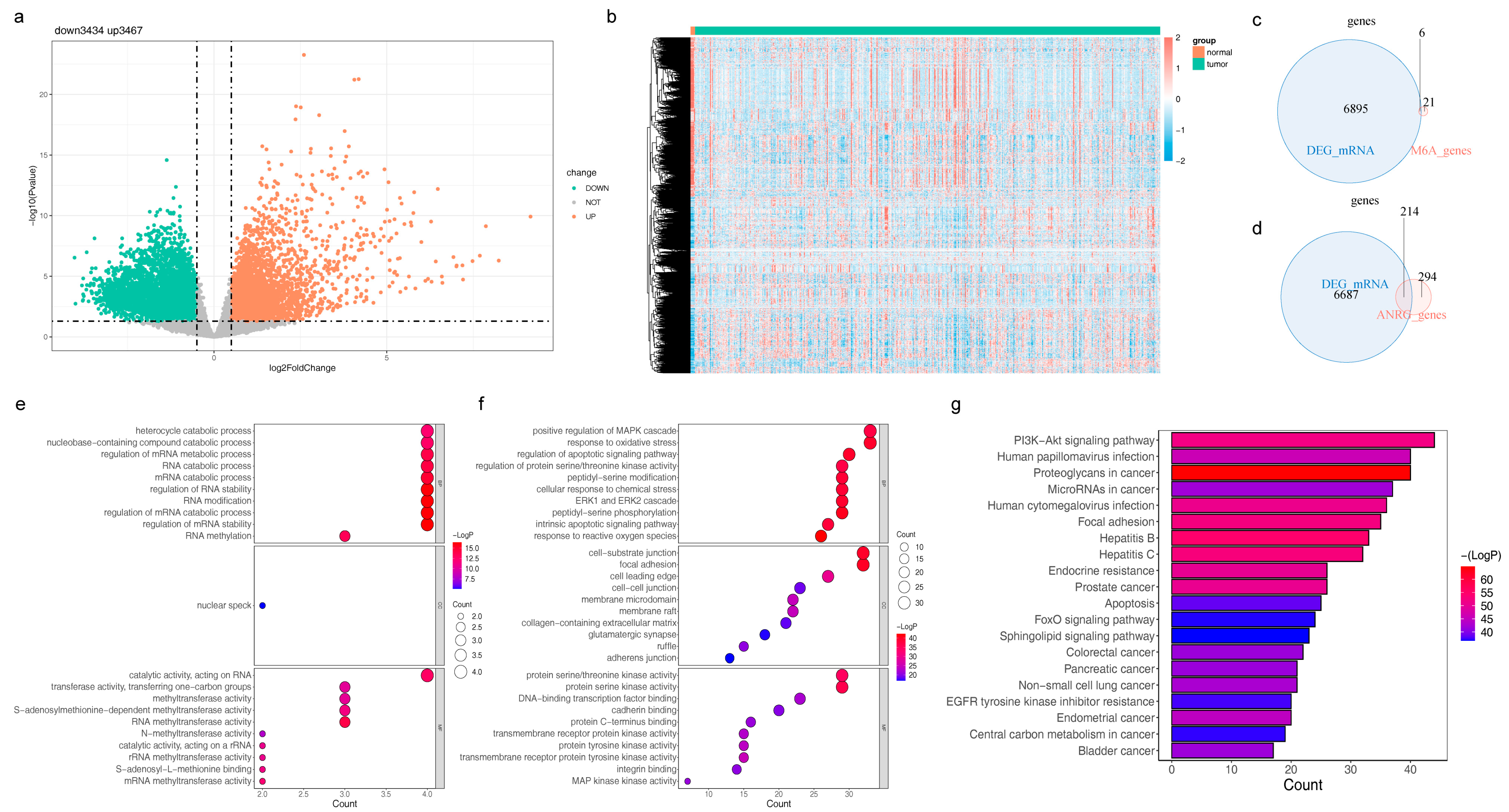
3.1. Identification and Functional Annotation Analysis of DE-m6A-RGs
3.2. Characterization of DE-m6A-ANRGs and Construction of the PPI Network
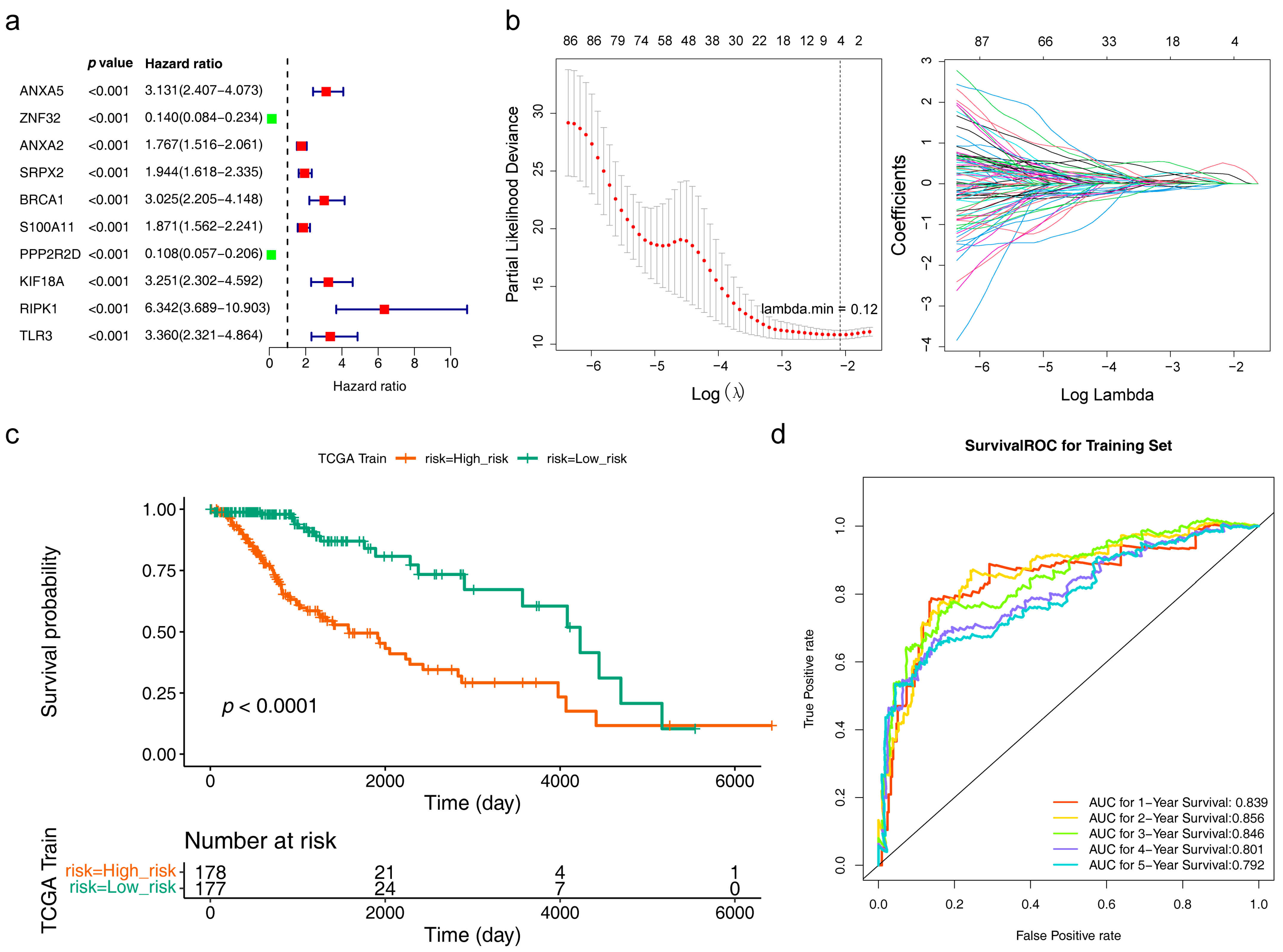
3.3. Acquisition and Assessment of the Risk Model
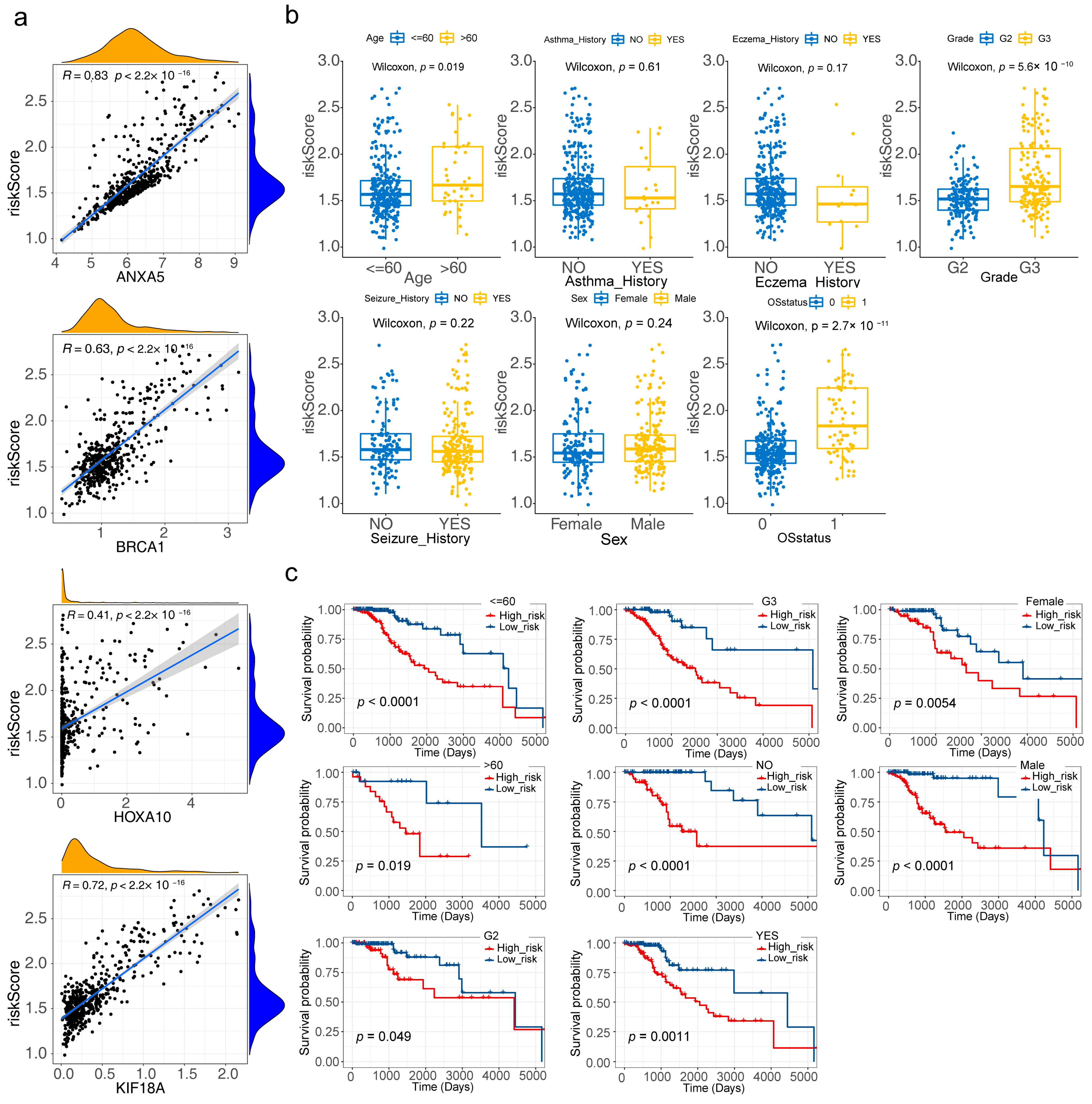
3.4. Risk Model Genes
3.5. Correlation Analysis of Clinical Characteristics
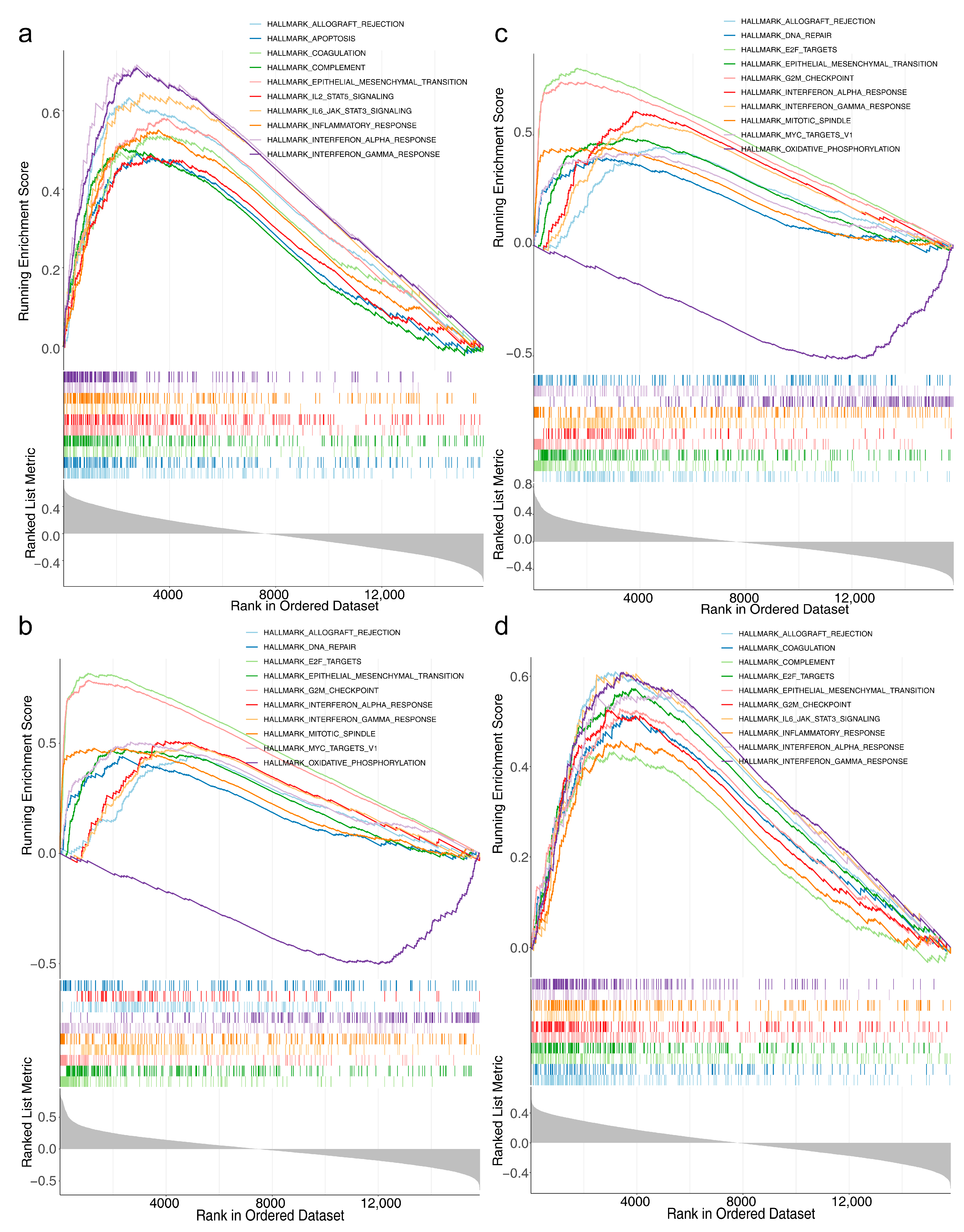
3.6. Acquisition of the Independent Prognostic Model and GSVA
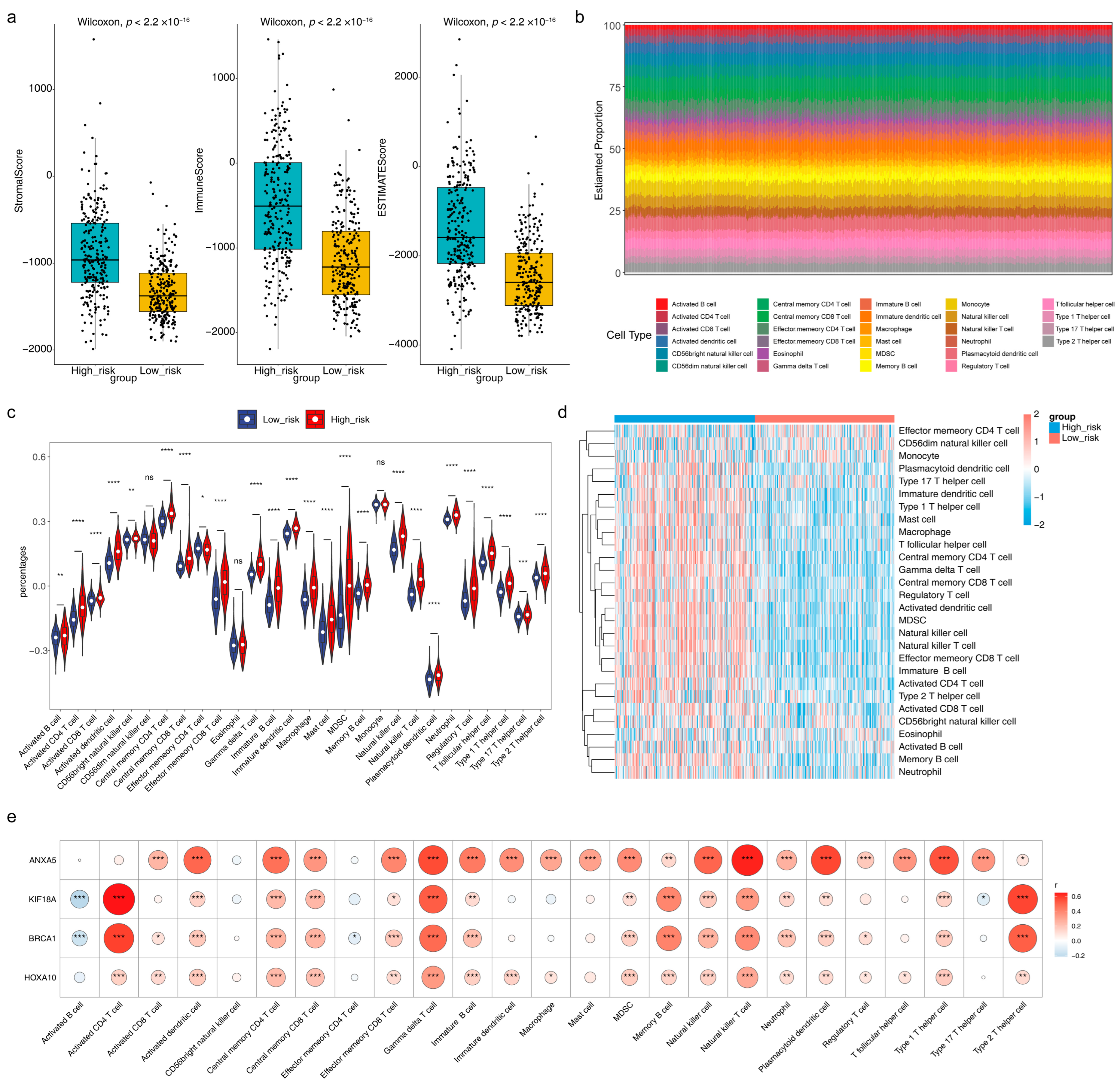
3.7. Immune Microenvironment Analysis and Prediction of Potential Therapeutic Agents
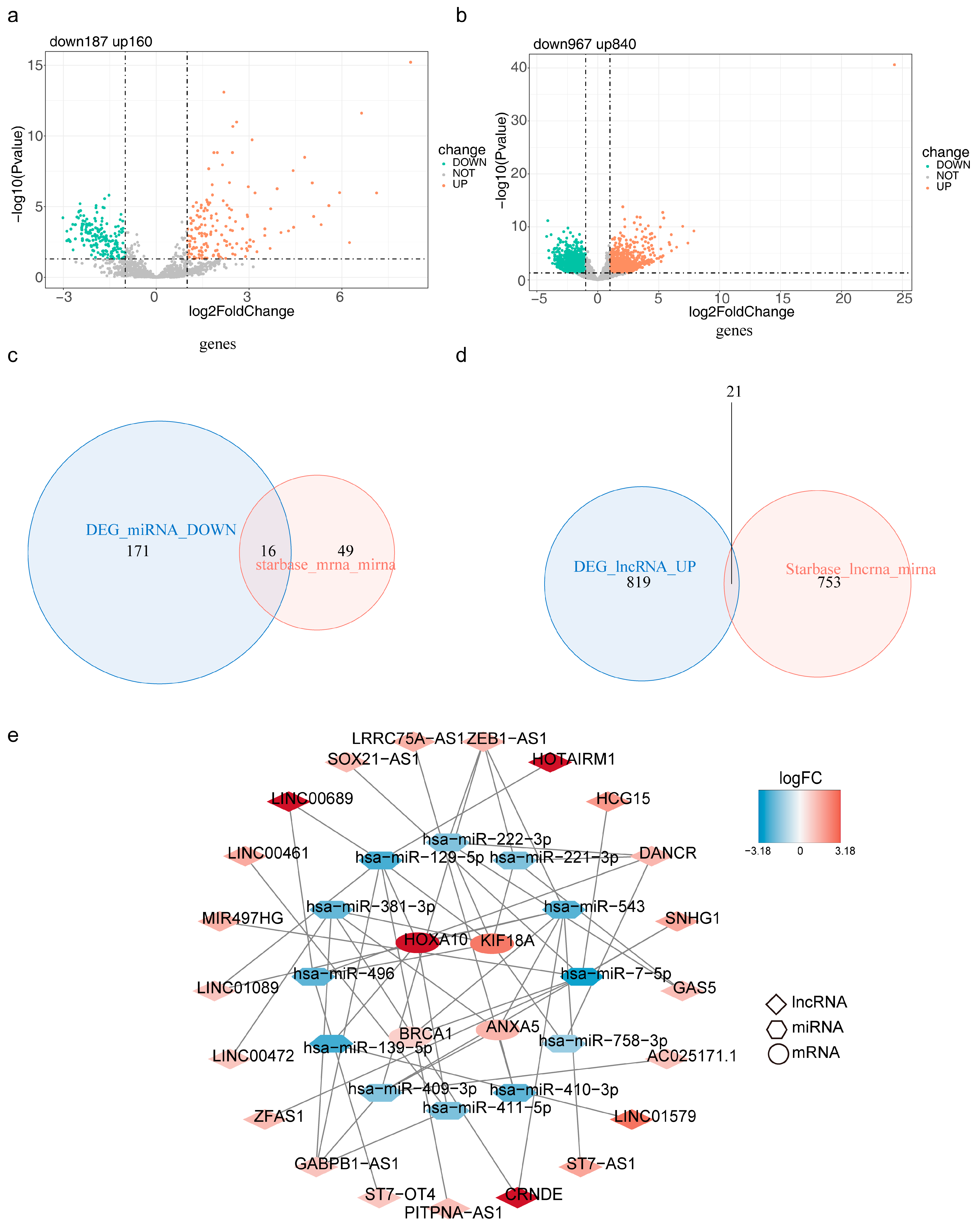
3.8. mRNA-miRNA-lncRNA Regulatory Network/
3.9. Expression of Risk Model Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| m6A | N6-methyladenosine |
| LGG | low-grade glioma |
| m6A-RGs | m6A-related genes |
| ANRGs | anoikis-related genes |
| DEGs | differentially expressed genes |
| LASSO | least absolute shrinkage and selection operator |
| GSEA | gene set enrichment analysis |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | protein interaction |
| ROC | receiver operating characteristic |
| K-M | Kaplan–Meier |
| GSVA | gene set variation analysis |
| IC50 | the 50% inhibitory concentration |
| ceRNA | competing endogenous RNA |
| HPA | human protein atlas |
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [PubMed]
- Morshed, R.A.; Young, J.S.; Hervey-Jumper, S.L.; Berger, M.S. The management of low-grade gliomas in adults. J. Neurosurg. Sci. 2019, 63, 450–457. [Google Scholar] [PubMed]
- Jansen, M.; Yip, S.; Louis, D.N. Molecular pathology in adult gliomas: Diagnostic, prognostic, and predictive markers. Lancet Neurol. 2010, 9, 717–726. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Krug, R.M.; Morgan, M.A.; Shatkin, A.J. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976, 20, 45–53. [Google Scholar]
- Du, J.; Ji, H.; Ma, S.; Jin, J.; Mi, S.; Hou, K.; Dong, J.; Wang, F.; Zhang, C.; Li, Y.; et al. m6A regulator-mediated methylation modification patterns and characteristics of immunity and stemness in low-grade glioma. Brief Bioinform. 2021, 22, bbab013. [Google Scholar]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar]
- Sanai, N.; Chang, S.; Berger, M.S. Low-grade gliomas in adults. J. Neurosurg. 2011, 115, 948–965. [Google Scholar]
- Zhu, Z.; Fang, C.; Xu, H.; Yuan, L.; Du, Y.; Ni, Y.; Xu, Y.; Shao, A.; Zhang, A.; Lou, M. Anoikis resistance in diffuse glioma: The potential therapeutic targets in the future. Front. Oncol. 2022, 12, 976557. [Google Scholar]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [PubMed]
- Deng, L.-J.; Deng, W.-Q.; Fan, S.-R.; Chen, M.-F.; Qi, M.; Lyu, W.-Y.; Qi, Q.; Tiwari, A.K.; Chen, J.-X.; Zhang, D.-M.; et al. m6A modification: Recent advances, anticancer targeted drug discovery and beyond. Mol. Cancer 2022, 21, 52. [Google Scholar] [PubMed]
- Chi, H.; Jiang, P.; Xu, K.; Zhao, Y.; Song, B.; Peng, G.; He, B.; Liu, X.; Xia, Z.; Tian, G. A novel anoikis-related gene signature predicts prognosis in patients with head and neck squamous cell carcinoma and reveals immune infiltration. Front. Genet. 2022, 13, 984273. [Google Scholar] [PubMed]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012, 16, 284–287. [Google Scholar]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21 (Suppl. 5), v1–v100. [Google Scholar]
- Yang, P.; Wang, Y.; Peng, X.; You, G.; Zhang, W.; Yan, W.; Bao, Z.; Wang, Y.; Qiu, X.; Jiang, T. Management and survival rates in patients with glioma in China (2004–2010): A retrospective study from a single-institution. J. Neurooncol. 2013, 113, 259–266. [Google Scholar]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar]
- Nguyen, Q.-H.; Nguyen, T.; Le, D.-H. Identification and Validation of a Novel Three Hub Long Noncoding RNAs With m6A Modification Signature in Low-Grade Gliomas. Front. Mol. Biosci. 2022, 9, 801931. [Google Scholar]
- Li, B.; Wang, F.; Wang, N.; Hou, K.; Du, J. Identification of Implications of Angiogenesis and m6A Modification on Immunosuppression and Therapeutic Sensitivity in Low-Grade Glioma by Network Computational Analysis of Subtypes and Signatures. Front. Immunol. 2022, 13, 871564. [Google Scholar]
- Zhao, S.; Chi, H.; Ji, W.; He, Q.; Lai, G.; Peng, G.; Zhao, X.; Cheng, C. A Bioinformatics-Based Analysis of an Anoikis-Related Gene Signature Predicts the Prognosis of Patients with Low-Grade Gliomas. Brain Sci. 2022, 12, 1349. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Tayara, H.; Zou, Q.; Chong, K.T. i6mA-Caps: A CapsuleNet-based framework for identifying DNA N6-methyladenine sites. Bioinformatics 2022, 38, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Körtel, N.; Rücklé, C.; Zhou, Y.; Busch, A.; Hoch-Kraft, P.; Sutandy, F.X.R.; Haase, J.; Pradhan, M.; Musheev, M.; Ostareck, D.; et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 2021, 49, e92. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xu, H.; Wu, Y.; Wang, J.; Yuan, Q. In silico genome-wide identification of m6A-associated SNPs as potential functional variants for periodontitis. J. Cell. Physiol. 2020, 235, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Guo, C.; Guan, H.; Liu, S.; Sun, M.-Z. Annexin A5 as a potential marker in tumors. Clin. Chim. Acta 2014, 427, 42–48. [Google Scholar] [CrossRef]
- Guo, X.; Song, J.; Zhao, J.; Wang, B.; Yang, Z.; Sun, P.; Hu, M. Impact of ANXA5 polymorphisms on glioma risk and patient prognosis. J. Neurooncol. 2019, 142, 11–26. [Google Scholar] [CrossRef]
- Ji, C.; Guo, H.; Zhang, P.; Kuang, W.; Fan, Y.; Wu, L. AnnexinA5 promote glioma cell invasion and migration via the PI3K/Akt/NF-κB signaling pathway. J. Neurooncol. 2018, 138, 469–478. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, S.; Kim, G.; Singh, P.; Kim, D.W. Integrative analysis of KIF4A, 9, 18A, and 23 and their clinical significance in low-grade glioma and glioblastoma. Sci. Rep. 2019, 9, 4599. [Google Scholar] [CrossRef]
- Tao, B.-Y.; Liu, Y.-Y.; Liu, H.-Y.; Zhang, Z.-H.; Guan, Y.-Q.; Wang, H.; Shi, Y.; Zhang, J. Prognostic Biomarker KIF18A and Its Correlations with Immune Infiltrates and Mitosis in Glioma. Front. Genet. 2022, 13, 852049. [Google Scholar] [CrossRef]
- Catteau, A.; Morris, J.R. BRCA1 methylation: A significant role in tumour development? Semin. Cancer Biol. 2002, 12, 359–371. [Google Scholar]
- Huang, T.; Chen, Y.; Zeng, Y.; Xu, C.; Huang, J.; Hu, W.; Chen, X.; Fu, H. Long non-coding RNA PSMA3-AS1 promotes glioma progression through modulating the miR-411-3p/HOXA10 pathway. BMC Cancer 2021, 21, 844. [Google Scholar] [CrossRef] [PubMed]
- Isaev, K.; Jiang, L.; Wu, S.; Lee, C.A.; Watters, V.; Fort, V.; Tsai, R.; Coutinho, F.J.; Hussein, S.M.I.; Zhang, J.; et al. Pan-cancer analysis of non-coding transcripts reveals the prognostic onco-lncRNA HOXA10-AS in gliomas. Cell Rep. 2021, 37, 109873. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, M.; Jia, M. Integrated Bioinformatic Analysis of the Correlation of HOXA10 Expression with Survival and Immune Cell Infiltration in Lower Grade Glioma. Biochem. Genet. 2023, 61, 238–257. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Ai, M.; Du, K.; Song, J.; Wang, B.; Lin, J.; Ren, A.; Chen, C.; Huang, Z.; Qiu, W.; et al. Pyroptosis relates to tumor microenvironment remodeling and prognosis: A pan-cancer perspective. Front. Immunol. 2022, 13, 1062225. [Google Scholar] [CrossRef]
- Narod, S.A. Which Genes for Hereditary Breast Cancer? N. Engl. J. Med. 2021, 384, 471–473. [Google Scholar] [CrossRef]
- Wang, B.; Cao, C.; Liu, X.; He, X.; Zhuang, H.; Wang, D.; Chen, B. BRCA1-associated protein inhibits glioma cell proliferation and migration and glioma stem cell self-renewal via the TGF-β/PI3K/AKT/mTOR signalling pathway. Cell. Oncol. 2020, 43, 223–235. [Google Scholar]
- Godfrey, D.I.; Le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar]
- Yue, C.; Yang, K.; Dong, W.; Hu, F.; Zhao, S.; Liu, S. γδ T Cells in Peripheral Blood of Glioma Patients. Med. Sci. Monit. 2018, 24, 1784–1792. [Google Scholar] [CrossRef]
- Rajendran, S.; Hu, Y.; Canella, A.; Peterson, C.; Gross, A.; Cam, M.; Nazzaro, M.; Haffey, A.; Serin-Harmanci, A.; Distefano, R.; et al. Single-cell RNA sequencing reveals immunosuppressive myeloid cell diversity during malignant progression in a murine model of glioma. Cell Rep. 2023, 42, 112197. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Oh, J.Y.; Okada, H.; Aghi, M.K. The immunology of low-grade gliomas. Neurosurg. Focus 2022, 52, E2. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Zhao, Y.; Zhou, H.; Tang, Y.; Xie, Z. The Comprehensive Analysis of m6A-Associated Anoikis Genes in Low-Grade Gliomas. Brain Sci. 2023, 13, 1311. https://doi.org/10.3390/brainsci13091311
Zheng H, Zhao Y, Zhou H, Tang Y, Xie Z. The Comprehensive Analysis of m6A-Associated Anoikis Genes in Low-Grade Gliomas. Brain Sciences. 2023; 13(9):1311. https://doi.org/10.3390/brainsci13091311
Chicago/Turabian StyleZheng, Hui, Yutong Zhao, Hai Zhou, Yuguang Tang, and Zongyi Xie. 2023. "The Comprehensive Analysis of m6A-Associated Anoikis Genes in Low-Grade Gliomas" Brain Sciences 13, no. 9: 1311. https://doi.org/10.3390/brainsci13091311
APA StyleZheng, H., Zhao, Y., Zhou, H., Tang, Y., & Xie, Z. (2023). The Comprehensive Analysis of m6A-Associated Anoikis Genes in Low-Grade Gliomas. Brain Sciences, 13(9), 1311. https://doi.org/10.3390/brainsci13091311