
Subacute Partially Reversible Leukoencephalopathy Expands the Aicardi–Goutières Syndrome Phenotype

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Data, Methods, and Materials
2.1. Participants’ Recruitment, Clinical Assessment, and Follow-Up
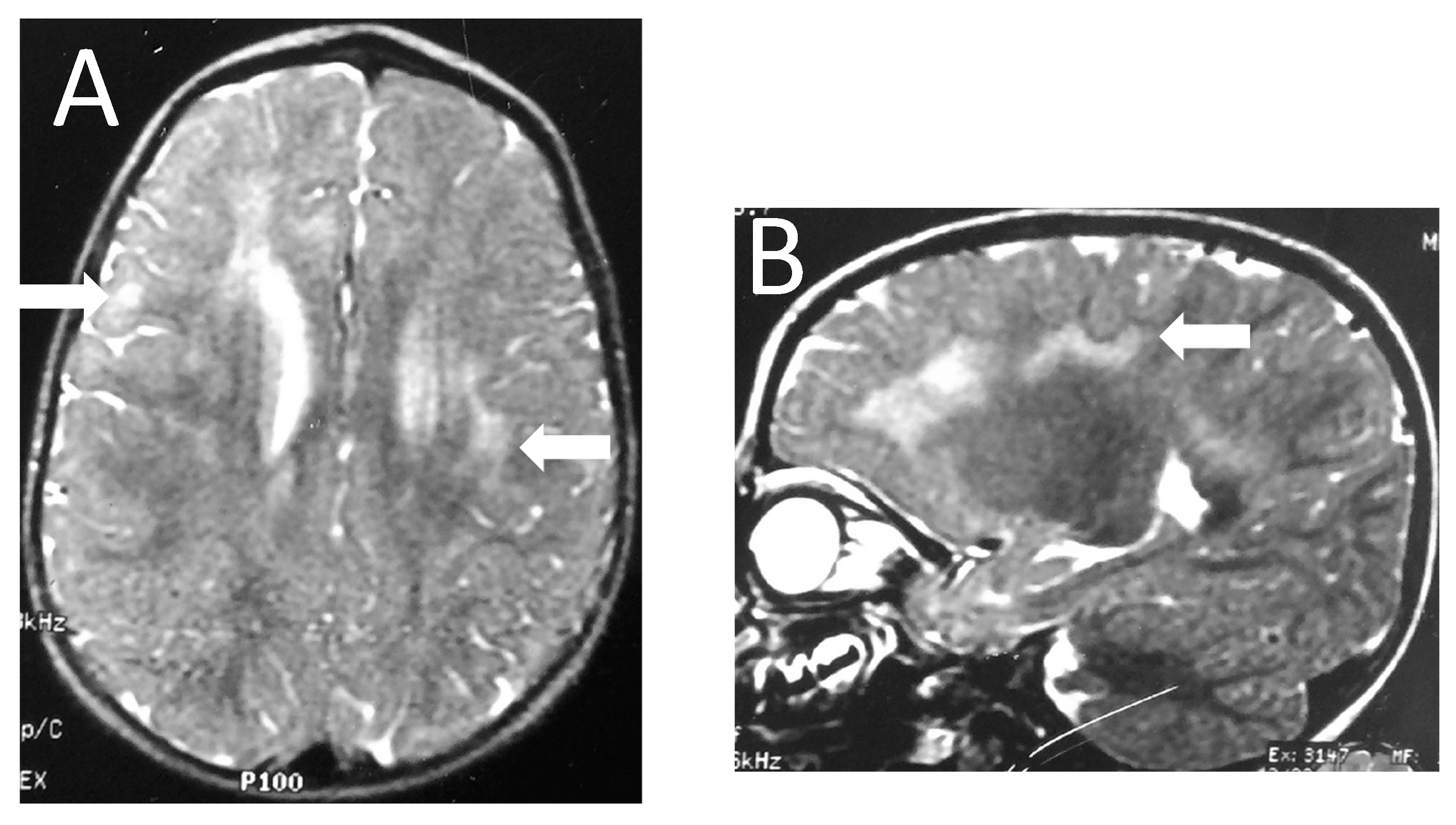
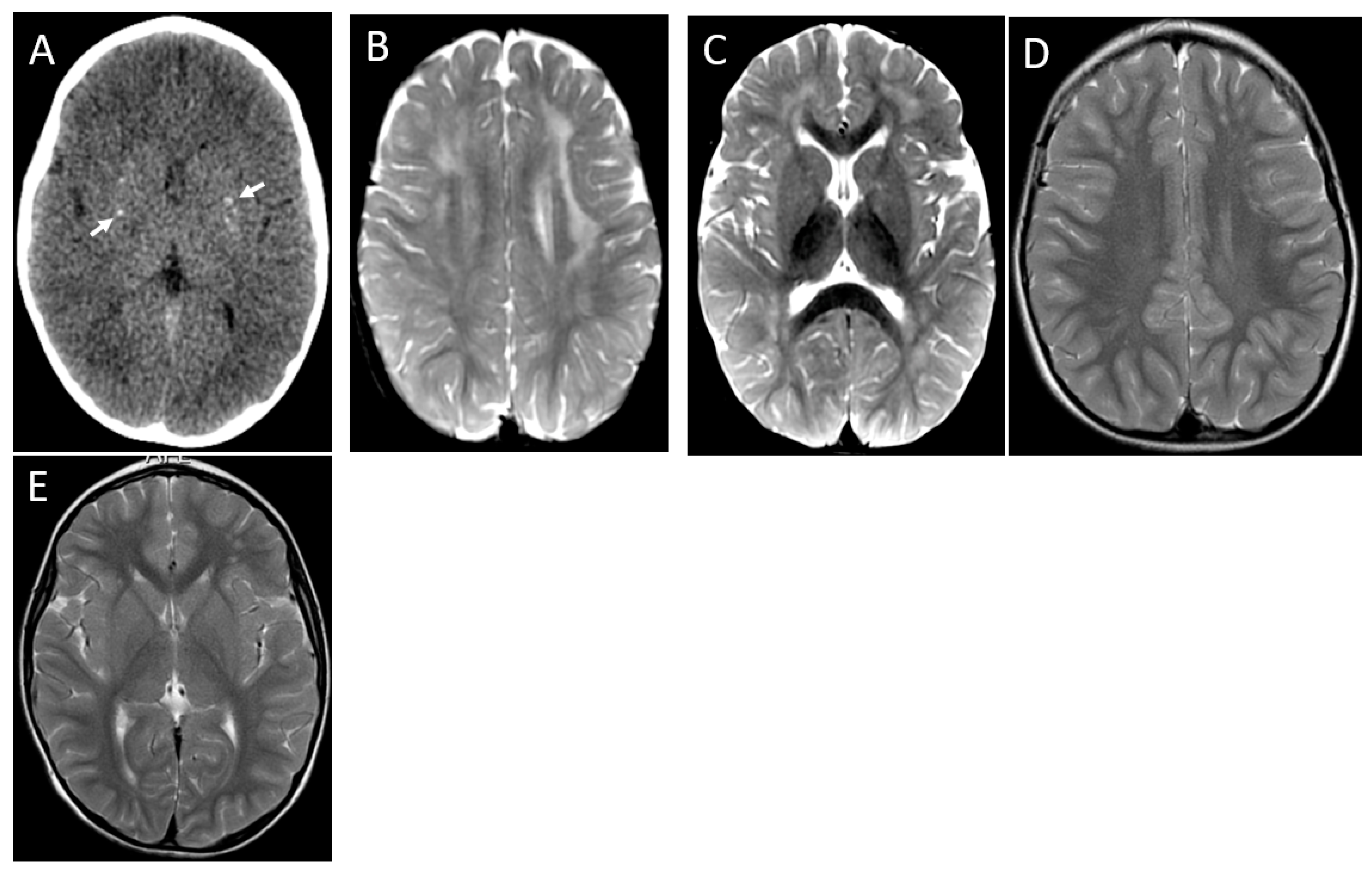
2.2. Neuroimaging
2.3. Laboratory
2.4. Molecular Analysis
2.5. Standard Protocol Approvals, Registrations, and Patient Consents
2.6. Data Availability Statement
3. Results
- Two individuals were homozygous for the variant p.Ala249Val in RNASEH2A. This rare missense substitution has never been reported in the literature at the time of the genetic test result, with a frequency at GnomAD [22] of 15 heterozygotes among 133,348 individuals (or ~1/9220). Both patients and their parents came from the same Northeastern Brazilian state of Ceara and are not known to be related. Alanine at codon 249 is highly conserved among biological species, and its substitution with valine is predicted to be deleterious by several computational programs (Polyphen, Provean, Mutation Taster).
- Three individuals were homozygous, and one was compound heterozygous for the variant p. Ala177Thr in RNASEH2B, with an allele frequency according to GnomAD [22] of 0.001361 (377 heterozygous for this variant among 138,512 individuals, or 1/367). In homozygosity or compound heterozygosity, this variant has been reported several times in the literature and is responsible for 90% of pathogenic alleles found in AGS2. The other variant was a rare nonsense mutation (p.Arg58*) never before reported in the literature and present in heterozygosity in only 2 of 138,535 individuals (or approximately 1/70,000).
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aicardi, J.; Goutières, F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann. Neurol. 1984, 15, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; La Piana, R.; Fazzi, E. Aicardi-Goutieres syndrome. Br. Med. Bull. 2009, 89, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Uggetti, C.; La Piana, R.; Orcesi, S.; Egitto, M.G.; Crow, Y.J.; Fazzi, E. Aicardi-Goutières syndrome: Neuroradiologic findings and follow-up. AJNR Am. J. Neuroradiol. 2009, 30, 1971–1976. [Google Scholar] [CrossRef]
- Vanderver, A.; Prust, M.; Kadom, N.; Demarest, S.; Crow, Y.J.; Helman, G.; Orcesi, S.; La Piana, R.; Uggetti, C.; Wang, J.; et al. Early onset Aicardi-Goutières syndrome: MRI pattern recognition. J. Child. Neurol. 2015, 30, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Lebon, P.; Badoual, J.; Ponsot, G.; Goutières, F.; Hémeury-Cukiert, F.; Aicardi, J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J. Neurol. Sci. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Crow, Y.J.; Livingston, J.H. Aicardi-Goutières syndrome: An important Mendelian mimic of congenital infection. Dev. Med. Child. Neurol. 2008, 50, 410–416. [Google Scholar] [CrossRef]
- Livingston, J.H.; Crow, Y.J. Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 and IFIH1: Aicardi-Goutières Syndrome and beyond. Neuropediatrics 2016, 47, 355–360. [Google Scholar]
- Orcesi, S.; Pessagno, A.; Biancheri, R.; La Piana, R.; Mascaretti, M.; Rossi, A.; Rice, G.; Crow, Y.; Fazzi, E.; Veneselli, E. Aicardi-Goutières syndrome presenting atypically as a sub-acute leukoencephalopathy. Eur. J. Paediatr. Neurol. 2008, 12, 408–411. [Google Scholar] [CrossRef]
- D’arrigo, S.; Riva, D.; Bulgheroni, S.; Chiapparini, L.; Lebon, P.; Rice, G.; Crow, Y.J.; Pantaleoni, C. Aicardi-Goutières syndrome: Description of a late onset case. Dev. Med. Child. Neurol. 2008, 50, 631–634. [Google Scholar] [CrossRef]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi- Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- Rice, G.I.; del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.A.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutières syndrome, and the type 1 interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- La Piana, R.; Uggetti, C.; Roncarolo, F.; Vanderver, A.; Olivieri, I.; Tonduti, D.; Helman, G.; Balottin, U.; Fazzi, E.; Crow, Y.J.; et al. Neuroradiologic patterns and novel imaging findings in Aicardi-Goutières syndrome. Neurology 2016, 86, 28–35. [Google Scholar] [CrossRef]
- Livingston, J.H.; Lin, J.P.; Dale, R.C.; Gill, D.; Brogan, P.; Munnich, A.; Kurian, M.A.; Gonzalez-Martinez, V.; De Goede, C.G.; Falconer, A.; et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J. Med. Genet. 2014, 51, 76–82. [Google Scholar] [CrossRef]
- La Piana, R.; Uggetti, C.; Olivieri, I.; Tonduti, D.; Balottin, U.; Fazzi, E.; Orcesi, S. Bilateral striatal necrosis in two subjects with Aicardi-Goutières syndrome due to mutations in ADAR1 (AGS6). Am. J. Med. Genet. A 2014, 164, 815–819. [Google Scholar] [CrossRef]
- Kothare, S.V.; Pungavkar, S.A.; Patkar, D.P.; Sainani, N.I.; Naik, M.H.; Gadani, S. Regression of white matter hypodensities with age in Aicardi- Goutierés syndrome: A case report. Childs Nerv. Syst. 2006, 22, 1503–1506. [Google Scholar] [CrossRef]
- La Piana, R.; Tran, L.; Guerrero, K.; Brais, B.; Levesque, S.; Sébire, G.; Riou, E.; Bernard, G. Spastic paraparesis and marked improvement of leukoencephalopathy in Aicardi-Goutières syndrome. Neuropediatrics 2014, 45, 406–410. [Google Scholar] [PubMed]
- Gnom, A.D. Available online: gnomad.broadinstitute.org/ (accessed on 9 May 2018).
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167, 296–312. [Google Scholar] [CrossRef] [PubMed]
- McEntagart, M.; Kamel, H.; Lebon, P.; King, M.D. Aicardi-Goutieres syndrome: An expanding phenotype. Neuropediatrics 1998, 29, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Polizzi, A.; Pavone, P.; Parano, E.; Incorpora, G.; Ruggieri, M. Lack of progression of brain atrophy in Aicardi-Goutières syndrome. Pediatr. Neurol. 2001, 24, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Ghezzi, D.; Haack, T.; Abbink, T.E.; Martinelli, D.; van Berkel, C.G.; Bley, A.; Diogo, L.; Grillo, E.; Naudé, J.T.W.; et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘TBL’ caused by EARS2 mutations. Brain 2012, 135 Pt 5, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Tenembaum, S.; Chamoles, N.; Fejerman, N. Acute disseminated encephalomyelitis: A long-term follow-up study of 84 pediatric patients. Neurology 2002, 59, 1224–1231. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Age at onset | 1 y 8 m Relapse at 5 y 8 m | 1 y 2 mo | 1 y 4 mo | 1 y 5 mo | 7 mo | 1 y 4 mo |
| Developmental condition before the onset | Walk without support Says several words | Walk without support Says at least 3 words | Walk without support Says at least 5 words | Walk without support Says at least 5 words | Sit without support | Walk without support Says at least 5 words |
| Age at first evaluation | 6 y | 9 y 2 mo | 8 y | 5 y | 21 y | 2 y 1 mo |
| Last clinical evaluation | 11 y | 14 y | 12 y | 9 y | 24 y | 6 y |
| Current age | 11 y | 14 y | 12 y | 10 y | 25 y | 6 y |
| Sex | M | F | M | M | M | M |
| Previous infection/immunization | Respiratory infection | Relapse-remitting fever | Multiple immunizations (PCV−10, TDp, Hib, and OPV) 10 days before the onset | Yellow fever vaccine, 7 days before the onset | Respiratory infection | Multiple immunizations (MMR, varicella, TDp, and OPV) 4 days before the onset |
| Consanguinity | No | Parents are first cousin | No | No | No | No |
| First clinical features | Ataxia Drowsiness Motor regression Spastic diplegia | Motor and language regression Spastic diplegia | Motor and language regression Spastic diplegia | Motor and language regression Spastic diplegia | Strabismus Seizure Motor regression Hypotonia | Motor and language regression Spastic diplegia |
| Time to reach the nadir of disease | 1st event: 1 mo 2nd event: a few days | 2 mo | 6 mo | 1 mo | 3 mo | 2 mo |
| Clinical features during the nadir of disease | Loss of head control Spastic diplegia | Loss of head control Speech loss Spastic diplegia | Loss of head control Speech loss Spastic diplegia Dystonia | Loss of head control Speech loss Spastic diplegia | Loss of head and sitting control | Loss of head control Speech loss Spastic diplegia |
| Treatment in the acute phase | Immunoglobulin in the 1st event | |||||
| IV steroid in both events | IV steroid | None | IV steroid | None | IV steroid | |
| Time to start milestones recovery | 3 mo in both | 1 y | 1 y | 1 y | 6 mo | 1 m |
| Actual clinical follow-up | Spastic diplegia Dysarthria Walk with support No concerns regarding cognition | Spastic diplegia Dysarthria Walk with support No concerns regarding cognition | Spastic diplegia Dysarthria Sit without support Dystonia No concerns regarding cognition | Spastic diplegia Dysarthria Walk with support No concerns regarding cognition | Spasticity of the right arm and left leg OCD Walk without support No concerns regarding cognition | Spastic diplegia Dysarthria Sit without support |
| CSF | 21 cells/mm3: 1st event 10 cells/mm3: 2nd event | 5 cells/mm3 | 8 cells/mm3 | 2 cells/mm3 | 4 cells/mm3 | 5 cells/mm3 |
| Molecular analysis | AGS2 RNASEH2B p.Gln58*/ p.Ala177Thr | AGS2 RNASEH2B p.Ala177Th/p.Ala177Thr | AGS4 RNASEH2A p.Ala249Val/p.Ala249Val | AGS2 RNASEH2B p.Ala177Thr/p.Ala177Thr | AGS4 RNASEH2A p.Ala249Va/ p.Ala249Val | AGS2 RNASEH2B p.Ala177Thr/p.Ala177Thr |
| GRCh37 (hg19) | chr13:51.503.646/ chr13:51.519.581 | chr13:51.519.581 | chr19:12.924.005 | chr13:51.519.581 | chr19:12.924.005 | chr13:51.519.581 |
| Image Modality | Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
| Distribution | Diffuse/homogeneous | Patchy/asymmetric | Patchy/asymmetric | Patchy/asymmetric | Patchy/asymmetric | Patchy/asymmetric | |
| MRI at onset | Predominant location | Diffuse | Frontal and anterior temporal | Frontal and anterior temporal | Frontal and anterior temporal | Frontal and anterior temporal | Frontal and parietal |
| Infratentorial involvement | Pons | - | - | - | - | Pons | |
| Contrast enhancement | + | - | - | - | - | - | |
| MRI at follow up | Months after onset | 1st episode 8 mo (2 y 3 mo old) | |||||
| 2nd episode | |||||||
| 12 mo | 4 y | 10 mo | 24 mo | 13 y | 5 mo | ||
| (6 years old) | (6 years old) | (2 y 2 mo old) | (3 y 2 mo old) | (13 years old) | (20 mo old) | ||
| WM lesions improvement | Marked | Marked | Marked | Marked | Marked | Marked | |
| Brain Atrophy | Mild | - | Mild | - | - | - | |
| CT scan | Time after onset | 5 y | 4 y | 7 y | 1 mo | 13 y | 2 mo |
| Calcification | - | - | - | Basal ganglia | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peixoto de Barcelos, I.; Bueno, C.; S. Godoy, L.F.; Pessoa, A.; A. Costa, L.; C. Monti, F.; Souza-Cabral, K.; Listik, C.; Castro, D.; Della-Ripa, B.; et al. Subacute Partially Reversible Leukoencephalopathy Expands the Aicardi–Goutières Syndrome Phenotype. Brain Sci. 2023, 13, 1169. https://doi.org/10.3390/brainsci13081169
Peixoto de Barcelos I, Bueno C, S. Godoy LF, Pessoa A, A. Costa L, C. Monti F, Souza-Cabral K, Listik C, Castro D, Della-Ripa B, et al. Subacute Partially Reversible Leukoencephalopathy Expands the Aicardi–Goutières Syndrome Phenotype. Brain Sciences. 2023; 13(8):1169. https://doi.org/10.3390/brainsci13081169
Chicago/Turabian StylePeixoto de Barcelos, Isabella, Clarissa Bueno, Luís Filipe S. Godoy, André Pessoa, Larissa A. Costa, Fernanda C. Monti, Katiane Souza-Cabral, Clarice Listik, Diego Castro, Bruno Della-Ripa, and et al. 2023. "Subacute Partially Reversible Leukoencephalopathy Expands the Aicardi–Goutières Syndrome Phenotype" Brain Sciences 13, no. 8: 1169. https://doi.org/10.3390/brainsci13081169
APA StylePeixoto de Barcelos, I., Bueno, C., S. Godoy, L. F., Pessoa, A., A. Costa, L., C. Monti, F., Souza-Cabral, K., Listik, C., Castro, D., Della-Ripa, B., Freua, F., C. Pires, L., T. Krüger, L., D. Gherpelli, J. L., B. Piazzon, F., P. Monteiro, F., T. Lucato, L., & Kok, F. (2023). Subacute Partially Reversible Leukoencephalopathy Expands the Aicardi–Goutières Syndrome Phenotype. Brain Sciences, 13(8), 1169. https://doi.org/10.3390/brainsci13081169