




Buffy Coat Score as a Biomarker of Treatment Response in Neuronal Ceroid Lipofuscinosis Type 2


Abstract
1. Introduction
2. Methods
2.1. Study Patients
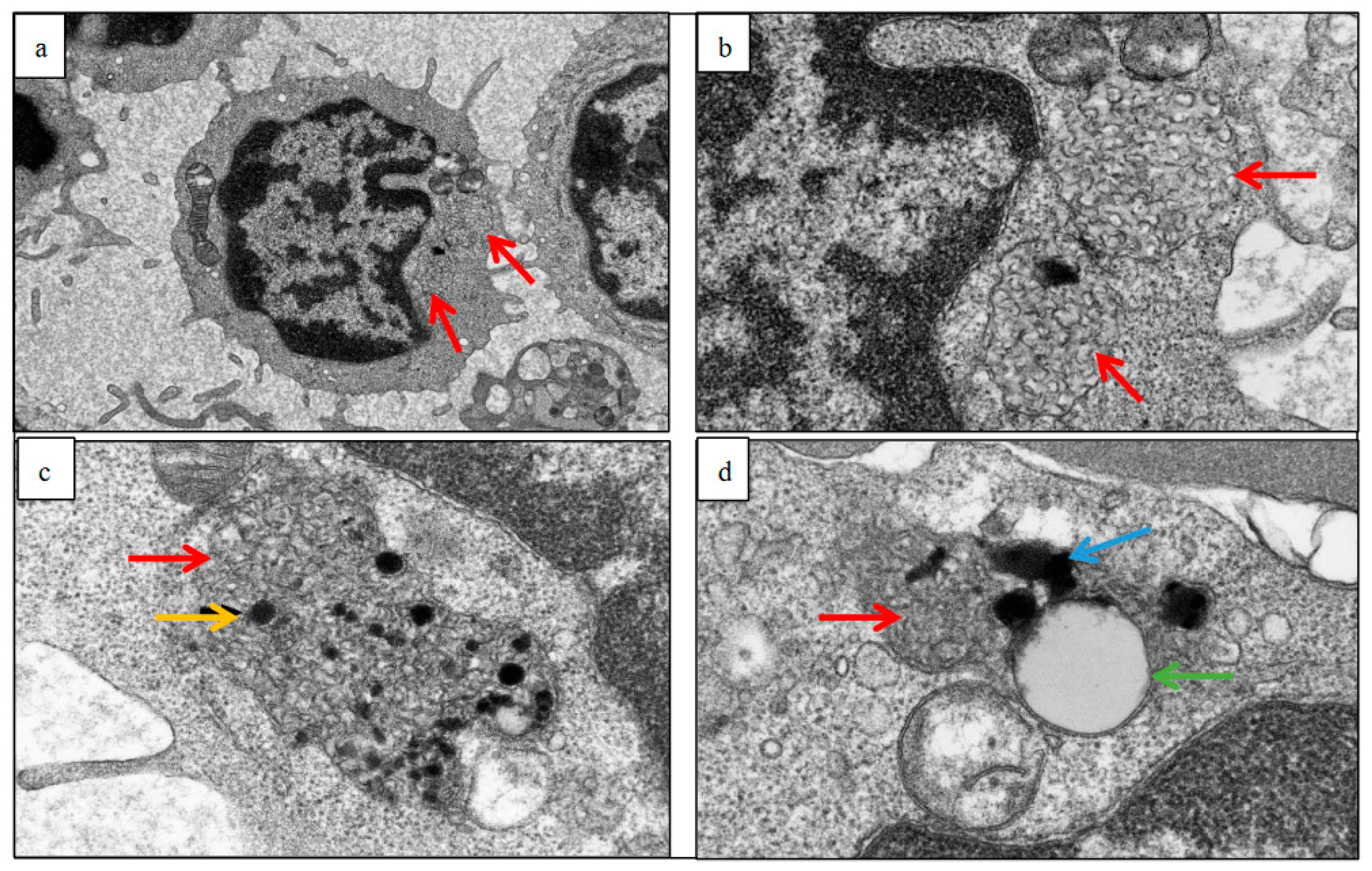
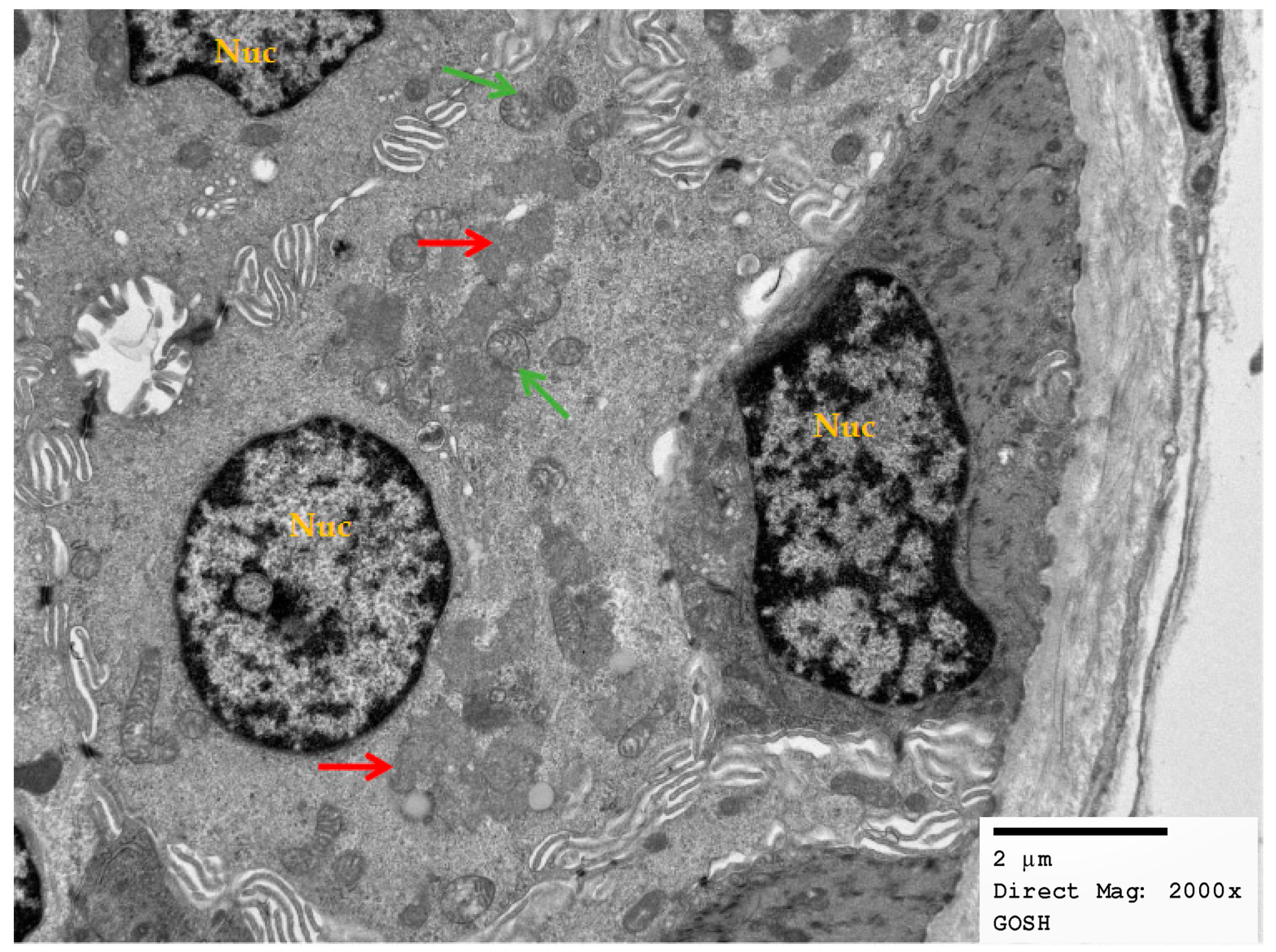
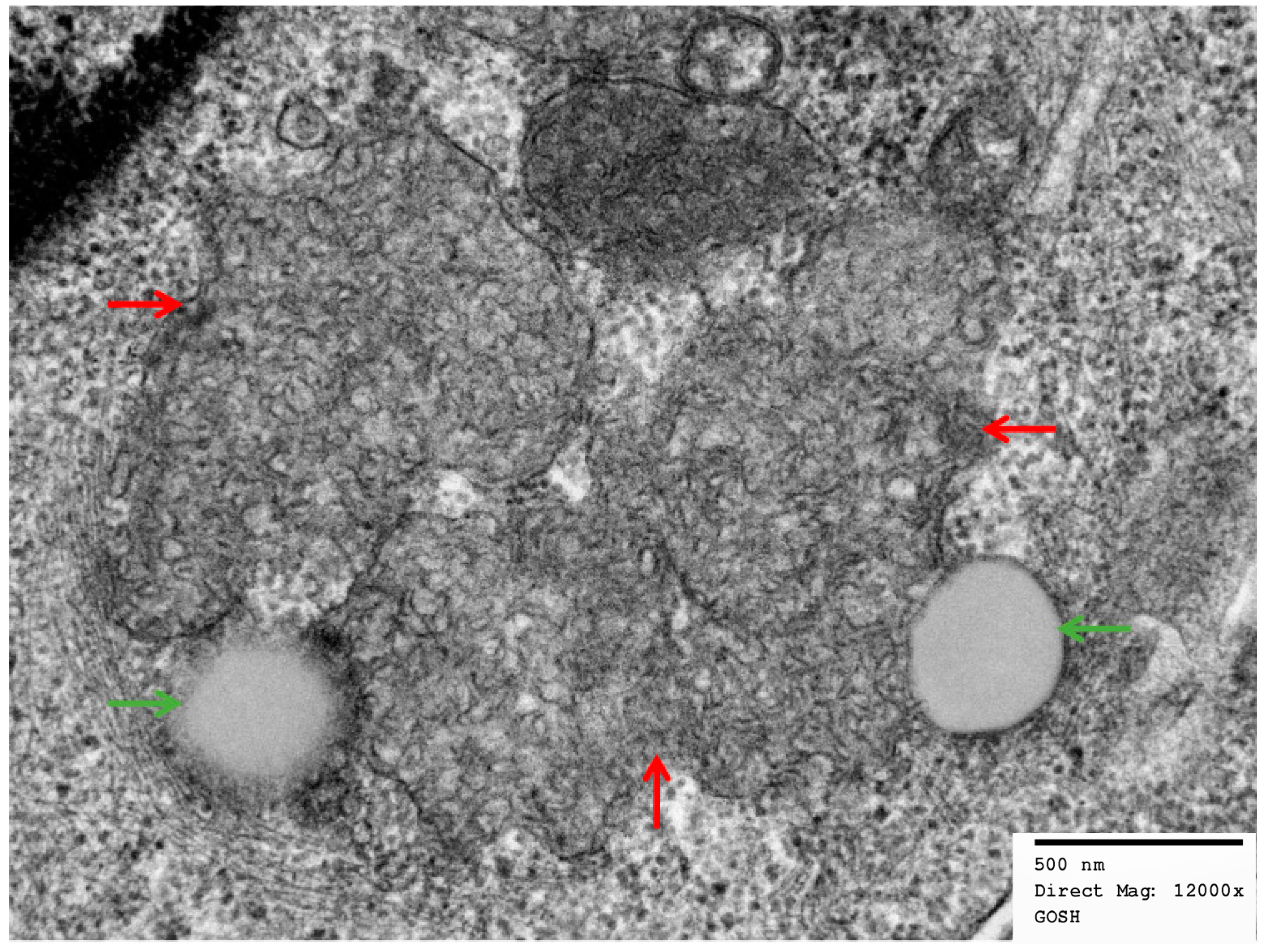
2.2. Electron Micrographs
2.3. Clinical Assessment
2.4. Statistics
3. Results
3.1. Demographics
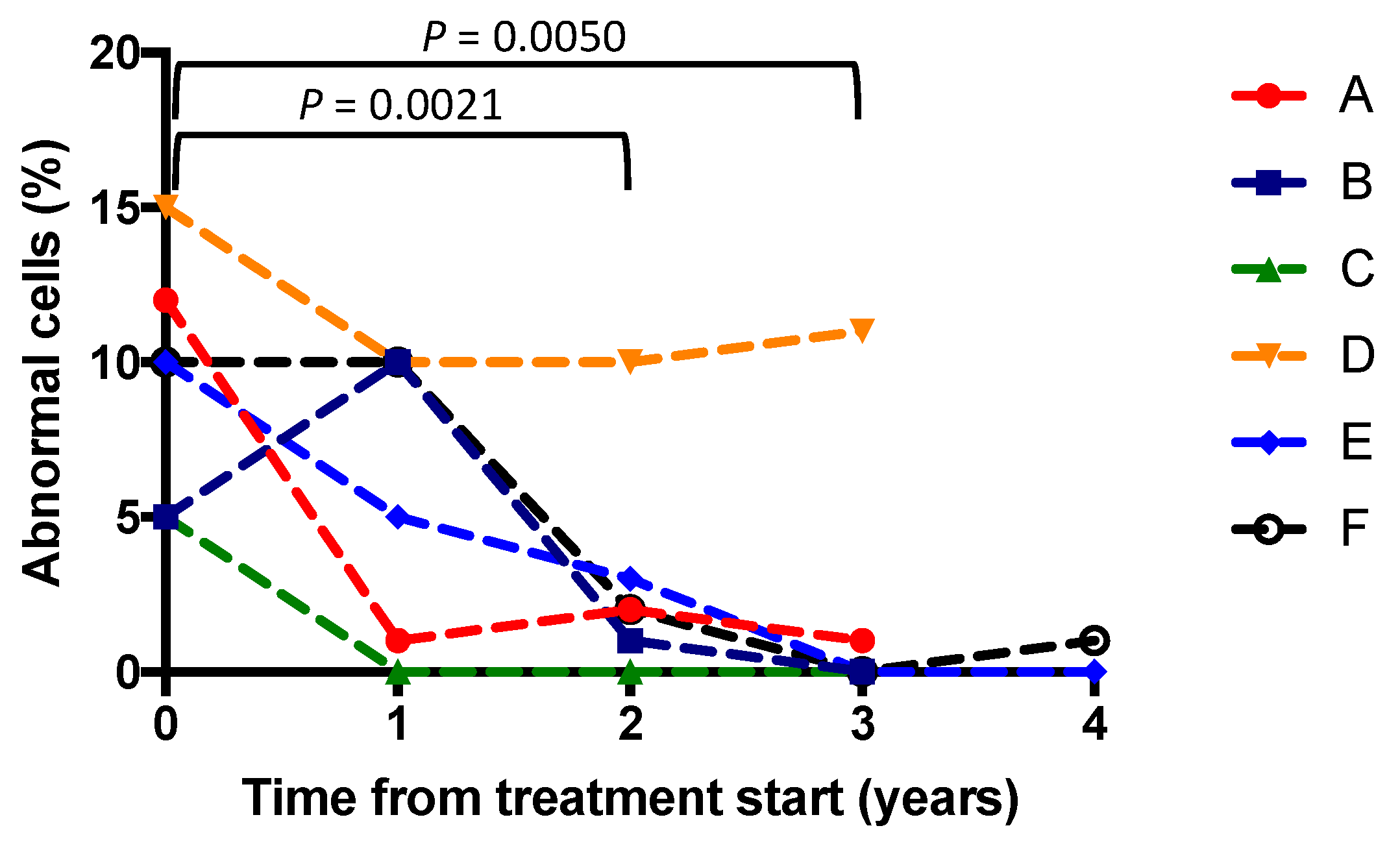
3.2. Buffy Coats
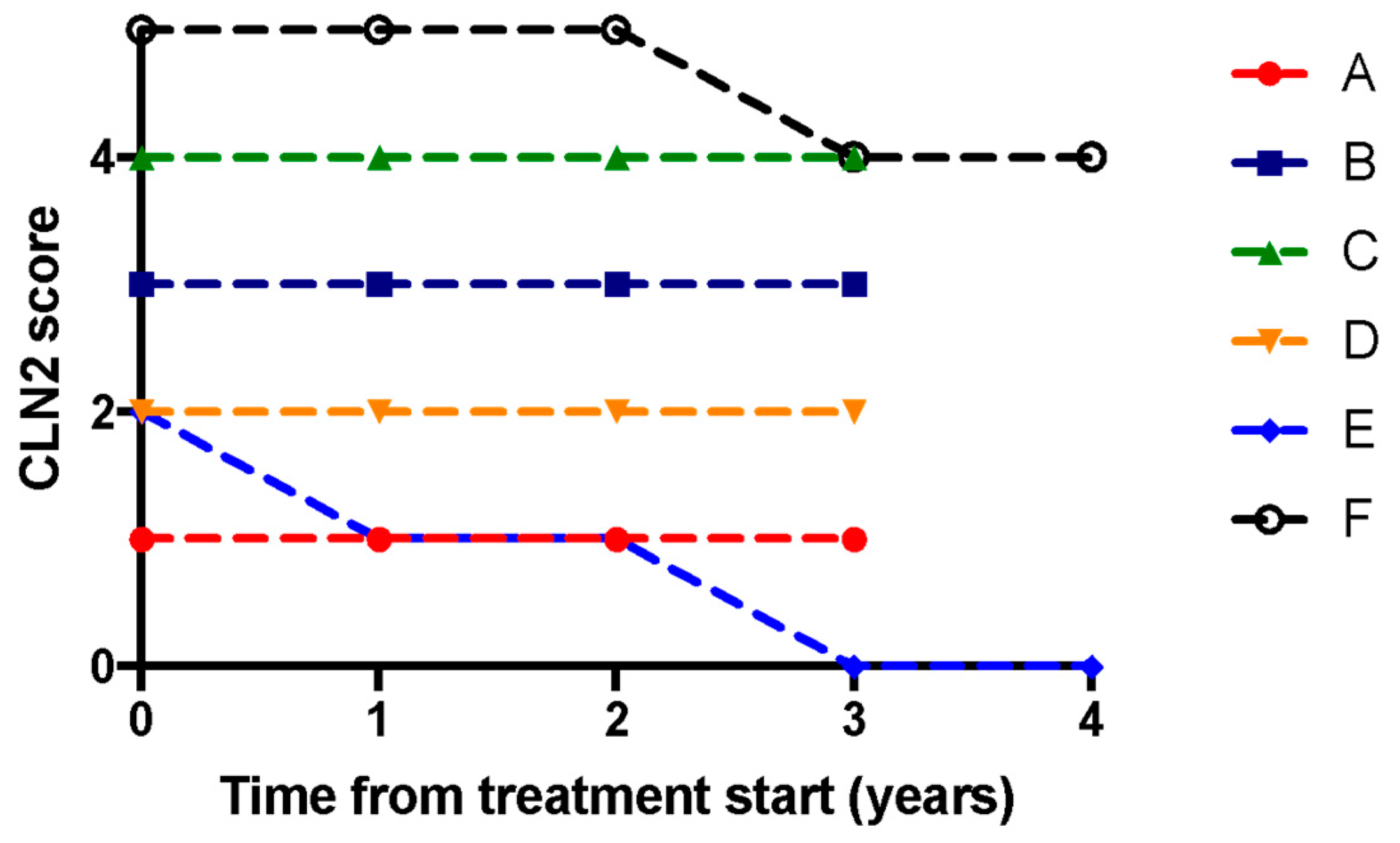
3.3. Clinical Outcomes
3.4. Skin Biopsies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mole, S.E.; Cotman, S.L. Genetics of the Neuronal Ceroid Lipofuscinoses (Batten Disease). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Warrier, V.; Vieira, M.; Mole, S.E. Genetic Basis and Phenotypic Correlations of the Neuronal Ceroid Lipofusinoses. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Kohlschütter, A.; Schulz, A. Towards Understanding the Neuronal Ceroid Lipofuscinoses. Brain Dev. 2009, 31, 499–502. [Google Scholar] [CrossRef]
- Moore, S.J.; Buckley, D.J.; Macmillan, A.; Marshall, H.D.; Steele, L.; Ray, P.N.; Nawaz, Z.; Baskin, B.; Frecker, M.; Carr, S.M.; et al. The Clinical and Genetic Epidemiology of Neuronal Ceroid Lipofuscinosis in Newfoundland. Clin. Genet. 2008, 74, 213–222. [Google Scholar] [CrossRef]
- Williams, R.E.; Adams, H.R.; Blohm, M.; Cohen-Pfeffer, J.L.; de los Reyes, E.; Denecke, J.; Drago, K.; Fairhurst, C.; Frazier, M.; Guelbert, N.; et al. Management Strategies for CLN2 Disease. Pediatr. Neurol. 2017, 69, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Donnelly, R.J.; Lackland, H.; Liu, C.G.; Sohar, I.; Pullarkat, R.K.; Lobel, P. Association of Mutations in a Lysosomal Protein with Classical Late- Infantile Neuronal Ceroid Lipofuscinosis. Science 1997, 277, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.A.; Wheeler, E.D.; Pearce, D.A. Screening, Diagnosis and Epidemiology of Batten Disease. Expert Opin. Orphan Drugs 2014, 2, 903–910. [Google Scholar] [CrossRef]
- Haltia, M. The Neuronal Ceroid-Lipofuscinoses. J. Neuropathol. Exp. Neurol. 2003, 62, 1–13. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between Genotype, Ultrastructural Morphology and Clinical Phenotype in the Neuronal Ceroid Lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL Neuropathology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2262–2266. [Google Scholar] [CrossRef]
- Mole, S.E.; Haltia, M. The Neuronal Ceroid-Lipofuscinoses (Batten Disease). In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 793–808. [Google Scholar] [CrossRef]
- Chang, M.; Cooper, J.D.; Davidson, B.L.; van Diggelen, O.P.; Elleder, M.; Goebel, H.H.; Golabek, A.A.; Kida, E.; Kohlschütter, A.; Lobel, P.; et al. CLN2. In The Neuronal Ceroid Lipofuscinoses (Batten Disease); Oxford University Press: Oxford, UK, 2011; pp. 80–109. [Google Scholar] [CrossRef]
- Schulz, A.; Kohlschütter, A.; Mink, J.; Simonati, A.; Williams, R. NCL Diseases—Clinical Perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Nickel, M.; Schulz, A. Natural History Studies in NCL and Their Expanding Role in Drug Development: Experiences from CLN2 Disease and Relevance for Clinical Trials. Front. Neurol. 2022, 13, 785841. [Google Scholar] [CrossRef]
- Gardner, E.; Bailey, M.; Schulz, A.; Aristorena, M.; Miller, N.; Mole, S.E. Mutation Update: Review of TPP1 Gene Variants Associated with Neuronal Ceroid Lipofuscinosis CLN2 Disease. Hum. Mutat. 2019, 40, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, C.M.; Pessoa, A.; Mendes, C.C.; Rivera-Nieto, C.; Vergara, D.; Troncoso, M.; Gardner, E.; Mallorens, F.; Tavera, L.; Lizcano, L.A.; et al. Revealing the Clinical Phenotype of Atypical Neuronal Ceroid Lipofuscinosis Type 2 Disease: Insights from the Largest Cohort in the World. J. Paediatr. Child Health 2020, 57, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Elleder, M.; Dvořáková, L.; Stolnaja, L.; Vlášková, H.; Hålková, H.; Druga, R.; Poupětová, H.; Košťálová, E.; Mikuláštík, J. Atypical CLN2 with Later Onset and Prolonged Course: A Neuropathologic Study Showing Different Sensitivity of Neuronal Subpopulations to TPP1 Deficiency. Acta Neuropathol. 2008, 116, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Kekatpure, M.V.; Heier, L.; Ballon, D.; Dyke, J.P.; Shungu, D.; Mao, X.; Kosofsky, B.; Kaplitt, M.G.; Souweidane, M.M.; et al. Neurological Deterioration in Late Infantile Neuronal Ceroid Lipofuscinosis. Neurology 2007, 69, 521–535. [Google Scholar] [CrossRef]
- Nickel, M.; Jacoby, D.; Lezius, S.; Down, M.; Simonati, A.; Genter, F.; Wittes, J.; Kohlschütter, A.; Schulz, A. Natural History of CLN2 Disease: Quantitative Assessment of Disease Characteristics and Rate of Progression. Neuropediatrics 2016, 47 (Suppl. S1), FV04-03. [Google Scholar] [CrossRef]
- Nickel, M.; Simonati, A.; Jacoby, D.; Lezius, S.; Kilian, D.; Van de Graaf, B.; Pagovich, O.E.; Kosofsky, B.; Yohay, K.; Downs, M.; et al. Disease Characteristics and Progression in Patients with Late-Infantile Neuronal Ceroid Lipofuscinosis Type 2 (CLN2) Disease: An Observational Cohort Study. Lancet Child Adolesc. Health 2018, 2, 582–590. [Google Scholar] [CrossRef]
- Specchio, N.; Bellusci, M.; Pietrafusa, N.; Trivisano, M.; de Palma, L.; Vigevano, F. Photosensitivity Is an Early Marker of Neuronal Ceroid Lipofuscinosis Type 2 Disease. Epilepsia 2017, 58, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Klein, K.A. Dementia in Childhood: Issues in Neuropsychological Assessment with Application to the Natural History and Treatment of Degenerative Storage Diseases. Adv. Child Neuropsychol. 1994, 1, 119–171. [Google Scholar] [CrossRef]
- Schulz, A.; Kohlschütter, A. NCL Disorders: Frequent Causes of Childhood Dementia. Iran. J. Child Neurol. 2013, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mink, J.W. Natural History Data for Childhood Neurodegenerative Disease. The Lancet Child and Adolescent Health. 2018, 2, 547–548. [Google Scholar] [CrossRef]
- Mole, S.E.; Schulz, A.; Badoe, E.; Berkovic, S.F.; de Los Reyes, E.C.; Dulz, S.; Gissen, P.; Guelbert, N.; Lourenco, C.M.; Mason, H.L.; et al. Guidelines on the Diagnosis, Clinical Assessments, Treatment and Management for CLN2 Disease Patients. Orphanet J. Rare Dis. 2021, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R.; Rasmussen, T.B.; Molgaard, H. Cardiac Involvement in Juvenile Neuronal Ceroid Lipofuscinosis (Batten Disease). Neurology 2011, 76, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, S.; Saito, Y.; Saito, T.; Komaki, H.; Nakagawa, E.; Sugai, K.; Sasaki, M.; Oka, A.; Takamisawa, I. Progressive Conduction Defects and Cardiac Death in Late Infantile Neuronal Ceroid Lipofuscinosis. Dev. Med. Child Neurol. 2011, 54, 663–666. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Trivisano, M. Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy. Ther. Clin. Risk Manag. 2020, 16, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz-Bełdzińska, M.; del Toro, M.; Haliloğlu, G.; Huidekoper, H.H.; Kravljanac, R.; Mühlhausen, C.; Andersen, B.N.; Prpić, I.; Striano, P.; Auvin, S. Managing CLN2 Disease: A Treatable Neurodegenerative Condition among Other Treatable Early Childhood Epilepsies. Expert Rev. Neurother. 2021, 21, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Labbé, E.E.; Lopez, I.; Murphy, L.; O’Brien, C. Optimism and Psychosocial Functioning in Caring for Children with Battens and Other Neurological Diseases. Psychol. Rep. 2002, 90, 1129. [Google Scholar] [CrossRef]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- Wibbeler, E.; Wang, R.; de los Reyes, E.; Specchio, N.; Gissen, P.; Guelbert, N.; Nickel, M.; Schwering, C.; Lehwald, L.; Trivisano, M.; et al. Cerliponase Alfa for the Treatment of Atypical Phenotypes of CLN2 Disease: A Retrospective Case Series. J. Child Neurol. 2020, 129, S161–S162. [Google Scholar] [CrossRef]
- Espitia Segura, O.M.; Hernández, Z.; Mancilla, N.I.; Naranjo, R.A.; Tavera, L. Real World Effectiveness of Cerliponase Alfa in Classical and Atypical Patients. A Case Series. Mol. Genet. Metab. Rep. 2021, 27, 100718. [Google Scholar] [CrossRef]
- Wyrwich, K.W.; Schulz, A.; Nickel, M.; Slasor, P.; Ajayi, T.; Jacoby, D.R.; Kohlschütter, A. An Adapted Clinical Measurement Tool for the Key Symptoms of CLN2 Disease. J. Inborn Errors Metab. Screen. 2018, 6, 232640981878838. [Google Scholar] [CrossRef]
- Bellettato, C.M.; Scarpa, M. Pathophysiology of Neuropathic Lysosomal Storage Disorders. J. Inherit. Metab. Dis. 2010, 33, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Smith, V.V.; Malone, M.; Sebire, N.J. Blood Film Examination for Vacuolated Lymphocytes in the Diagnosis of Metabolic Disorders; Retrospective Experience of More than 2500 Cases from a Single Centre. J. Clin. Pathol. 2005, 58, 1305–1310. [Google Scholar] [CrossRef]
- Bajel, A.; Bazargan, A.; Doery, J.; Roy, S. Lymphocyte Vacuolation: Clue to Inherited Metabolic Disease. Pathology 2010, 42, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Baumann, R.J.; Markesbery, W.R. Juvenile Amaurotic Idiocy (Neuronal Ceroid Lipofuscinosis) and Lymphocyte Fingerprint Profiles. Ann. Neurol. 1978, 4, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Smith, V.V.; Brooke, I.; Malone, M.; Sebire, N.J. Diagnosis of Neuronal Ceroid Lipofuscinosis (Batten Disease) by Electron Microscopy in Peripheral Blood Specimens. Ultrastruct. Pathol. 2006, 30, 373–378. [Google Scholar] [CrossRef]
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human Pathology in NCL. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1807–1826. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Late-Infantile Neuronal Ceroid-Lipofuscinosis. Arch. Neurol. 1976, 33, 630. [Google Scholar] [CrossRef]
- Vogler, C.; Rosenberg, H.S.; Williams, J.C.; Butler, I. Electron Microscopy in the Diagnosis of Lysosomal Storage Diseases. Am. J. Med. Genet. Suppl. 1987, 3, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Zeman, W.; Dyken, P. Neuronal Ceroid-Lipofuscinosis (Batten’s Disease): Relationship to Amaurotic Family Idiocy? Pediatrics 1969, 44, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Fietz, M.; AlSayed, M.; Burke, D.; Cohen-Pfeffer, J.; Cooper, J.D.; Dvořáková, L.; Giugliani, R.; Izzo, E.; Jahnová, H.; Lukacs, Z.; et al. Diagnosis of Neuronal Ceroid Lipofuscinosis Type 2 (CLN2 Disease): Expert Recommendations for Early Detection and Laboratory Diagnosis. Mol. Genet. Metab. 2016, 119, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Katz, M.L.; Coates, J.R.; Sibigtroth, C.M.; Taylor, J.D.; Carpentier, M.; Young, W.M.; Wininger, F.A.; Kennedy, D.; Vuillemenot, B.R.; O’Neill, C.A. Enzyme Replacement Therapy Attenuates Disease Progression in a Canine Model of Late-infantile Neuronal Ceroid Lipofuscinosis (CLN2 Disease). J. Neurosci. Res 2014, 92, 1591–1598. [Google Scholar] [CrossRef]
- Schwering, C.; Kammler, G.; Wibbeler, E.; Christner, M.; Knobloch, J.K.-M.; Nickel, M.; Denecke, J.; Baehr, M.; Schulz, A. Development of the “Hamburg Best Practice Guidelines for ICV−Enzyme Replacement Therapy (ERT) in CLN2 Disease” Based on 6 Years Treatment Experience in 48 Patients. J. Child Neurol. 2021, 36, 635–641. [Google Scholar] [CrossRef]
- Fagerland, J.A. Preparation of Buffy Coat Leukocytes for Transmission Electron Microscopy. Microsc. Today 1999, 7, 29. [Google Scholar] [CrossRef]
- Choudhary, O.P.; Sarkar, R.; Priyanka; Chethan, G.E.; Doley, P.J.; Chandra Kalita, P.; Kalita, A. Preparation of Blood Samples for Electron Microscopy: The Standard Protocol. Ann. Med. Surg. 2021, 70, 102895. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.J.; Klumperman, J. Electron Microscopy in Cell Biology: Integrating Structure and Function. Nat. Rev. Mol. Cell Biol. 2003, 4, SS6–SS9. Available online: https://pubmed.ncbi.nlm.nih.gov/14587520/ (accessed on 5 January 2023).
- Kuper, W.F.E.; Oostendorp, M.; van den Broek, B.T.A.; van Veghel, K.; Nonkes, L.J.P.; Nieuwenhuis, E.E.S.; Fuchs, S.A.; Veenendaal, T.; Klumperman, J.; Huisman, A.; et al. Quantifying Lymphocyte Vacuolization Serves as a Measure of CLN3 Disease Severity. JIMD Rep. 2020, 54, 87–97. [Google Scholar] [CrossRef]
- Dolman, C.L.; MacLeod, P.M.; Chang, E. Skin Punch Biopsies and Lymphocytes in the Diagnosis of Lipidoses. Can. J. Neurol. Sci. 1975, 2, 67–73. [Google Scholar] [CrossRef]
- Dunnett, C.W. A Multiple Comparison Procedure for Comparing Several Treatments with a Control. J. Am. Stat. Assoc. 1955, 50, 1096–1121. [Google Scholar] [CrossRef]
- Friedman, M. The Use of Ranks to Avoid the Assumption of Normality Implicit in the Analysis of Variance. J. Am. Stat. Assoc. 1937, 32, 675–701. [Google Scholar] [CrossRef]
- Wisniewski, K.E. The Diagnostic Value of Ultrastructural Studies of Skin-Punch Biopsies and Buffy Coat for the Early Diagnosis of Some Neurodegenerative Diseases. Ann. N. Y. Acad. Sci. 1986, 477, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.J.; de Groote, C. Involvement of the Skin in Late Infantile and Juvenile Amaurotic Idiocies (Neuronal Ceroid-Lipofuscinoses). Pathol. Eur. 1974, 9, 263–272. Available online: https://europepmc.org/article/MED/4457780 (accessed on 6 January 2023).
- Steinfeld, R.; Heim, P.; Von Gregory, H.; Meyer, K.; Ullrich, K.; Goebel, H.H.; Kohlschütter, A. Late Infantile Neuronal Ceroid Lipofuscinosis: Quantitative Description of the Clinical Course in Patients with CLN2 Mutations. Am. J. Med. Genet. 2002, 112, 347–354. [Google Scholar] [CrossRef]
- Papandreou, A.; Doykov, I.; Spiewak, J.; Komarov, N.; Habermann, S.; Kurian, M.A.; Mills, P.B.; Mills, K.; Gissen, P.; Heywood, W.E. Niemann–Pick Type C Disease as Proof-of-concept for Intelligent Biomarker Panel Selection in Neurometabolic Disorders. Dev. Med. Child Neurol. 2022, 64, 1539–1546. [Google Scholar] [CrossRef]
- Baker, E.H.; Levin, S.W.; Zhang, Z.; Mukherjee, A.B. MRI Brain Volume Measurements in Infantile Neuronal Ceroid Lipofuscinosis. Am. J. Neuroradiol. 2017, 38, 376–382. [Google Scholar] [CrossRef]
- Todiere, G.; della Vecchia, S.; Morales, M.A.; Barison, A.; Ricca, I.; Tessa, A.; Colombi, E.; Santorelli, F.M. Cardiac Magnetic Resonance Findings in Neuronal Ceroid Lipofuscinosis: A Case Report. Front. Neurol. 2022, 13, 942667. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Swier, V.J.; Johnson, T.B.; Cain, J.C.; Pratt, M.; Rechtzigel, M.; Leppert, H.; Dang Do, A.N.; Porter, F.D.; Weimer, J.M. Glycerophosphoinositol Is Elevated in Blood Samples from CLN3 Δex7–8 Pigs, Cln3 Δex7–8 Mice, and CLN3-Affected Individuals. Biomark. Insights 2022, 17, 117727192211077. [Google Scholar] [CrossRef]
- Iwan, K.; Patel, N.; Heslegrave, A.; Borisova, M.; Lee, L.; Bower, R.; Mole, S.E.; Mills, P.B.; Zetterberg, H.; Mills, K.; et al. Cerebrospinal Fluid Neurofilament Light Chain Levels in CLN2 Disease Patients Treated with Enzyme Replacement Therapy Normalise after Two Years on Treatment. F1000Res 2022, 10, 614. [Google Scholar] [CrossRef]
- Ru, Y.; Corado, C.; Soon, R.K.; Melton, A.C.; Harris, A.; Yu, G.K.; Pryer, N.; Sinclair, J.R.; Katz, M.L.; Ajayi, T.; et al. Neurofilament Light Is a Treatment-responsive Biomarker in CLN2 Disease. Ann. Clin. Transl. Neurol. 2019, 6, 2437–2447. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Current Age | Age at Diagnosis | Mutation | Age at Treatment Start | CLN2 Scores at Start of Treatment (Motor, Language) |
|---|---|---|---|---|---|
| A | 10 years 2 months | 4 years 7 months | c.89 + 5G > A, c 509. 1G > C | 4 years 10 months | 1, 0 |
| B | 10 years 1 months | 4 years 4 months | homozygous c.509-1G > C | 4 years 7 months | 1, 2 |
| C | 20 years 3 months | 13 years 3 months | c.89 + 5G > C c. 1340G > A | 14 years 11 months | 2, 2 |
| D | 9 years 4 months | 4 years 2 months | homozygous c.1052 G > T | 4 years 5 months | 2, 0 |
| E | 11 years 4 months | 4 years | homozygous c.1052 G > T | 5 years 10 months | 1, 1 |
| F | 9 years 2 months | 2 years 2 months | homozygous c.1052 G > T | 3 years 11 months | 3, 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivananthan, S.; Lee, L.; Anderson, G.; Csanyi, B.; Williams, R.; Gissen, P. Buffy Coat Score as a Biomarker of Treatment Response in Neuronal Ceroid Lipofuscinosis Type 2. Brain Sci. 2023, 13, 209. https://doi.org/10.3390/brainsci13020209
Sivananthan S, Lee L, Anderson G, Csanyi B, Williams R, Gissen P. Buffy Coat Score as a Biomarker of Treatment Response in Neuronal Ceroid Lipofuscinosis Type 2. Brain Sciences. 2023; 13(2):209. https://doi.org/10.3390/brainsci13020209
Chicago/Turabian StyleSivananthan, Siyamini, Laura Lee, Glenn Anderson, Barbara Csanyi, Ruth Williams, and Paul Gissen. 2023. "Buffy Coat Score as a Biomarker of Treatment Response in Neuronal Ceroid Lipofuscinosis Type 2" Brain Sciences 13, no. 2: 209. https://doi.org/10.3390/brainsci13020209
APA StyleSivananthan, S., Lee, L., Anderson, G., Csanyi, B., Williams, R., & Gissen, P. (2023). Buffy Coat Score as a Biomarker of Treatment Response in Neuronal Ceroid Lipofuscinosis Type 2. Brain Sciences, 13(2), 209. https://doi.org/10.3390/brainsci13020209