
A New Case of Autosomal-Dominant POLR3B-Related Disorder: Widening Genotypic and Phenotypic Spectrum

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case Description
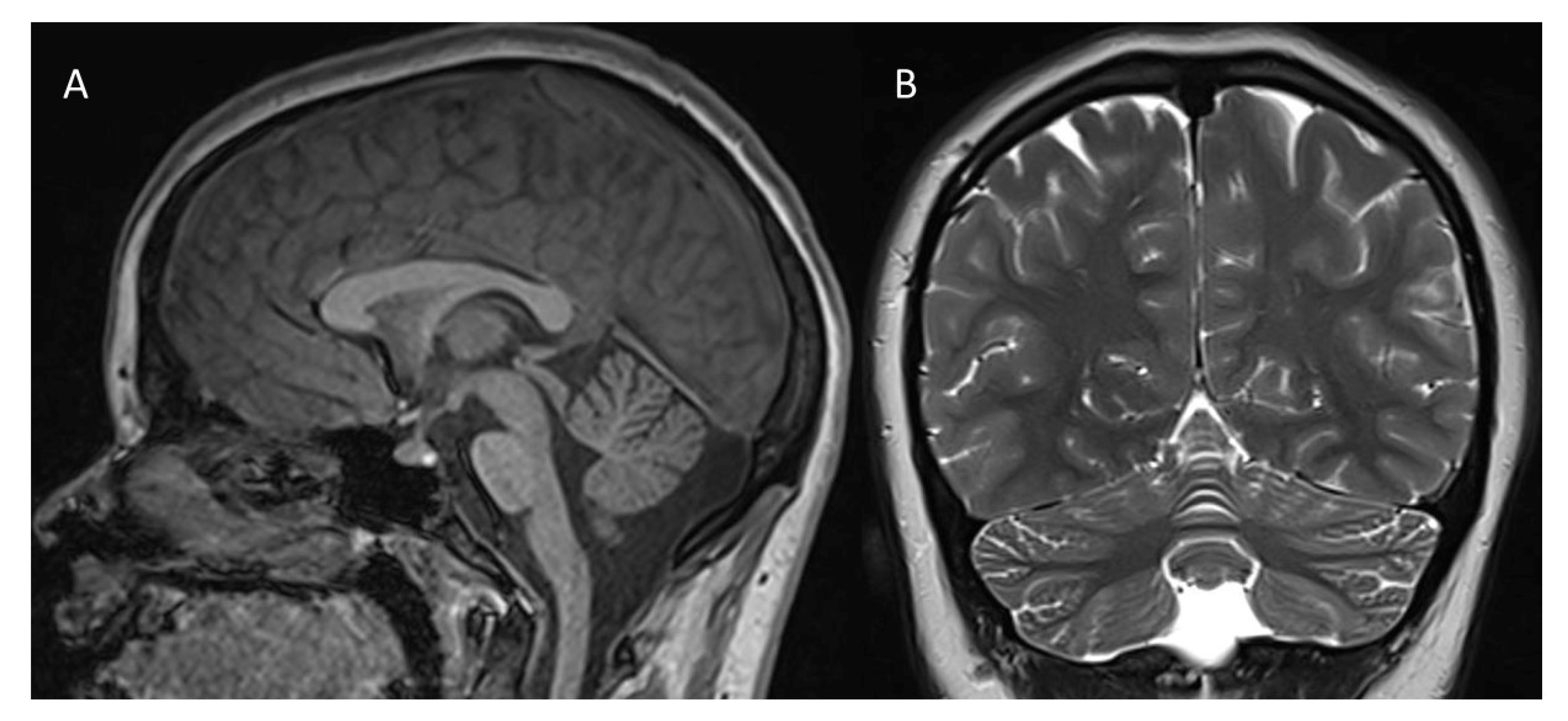
2.1. Clinical Report
2.2. Genetics Results
2.3. Methods
3. Discussion
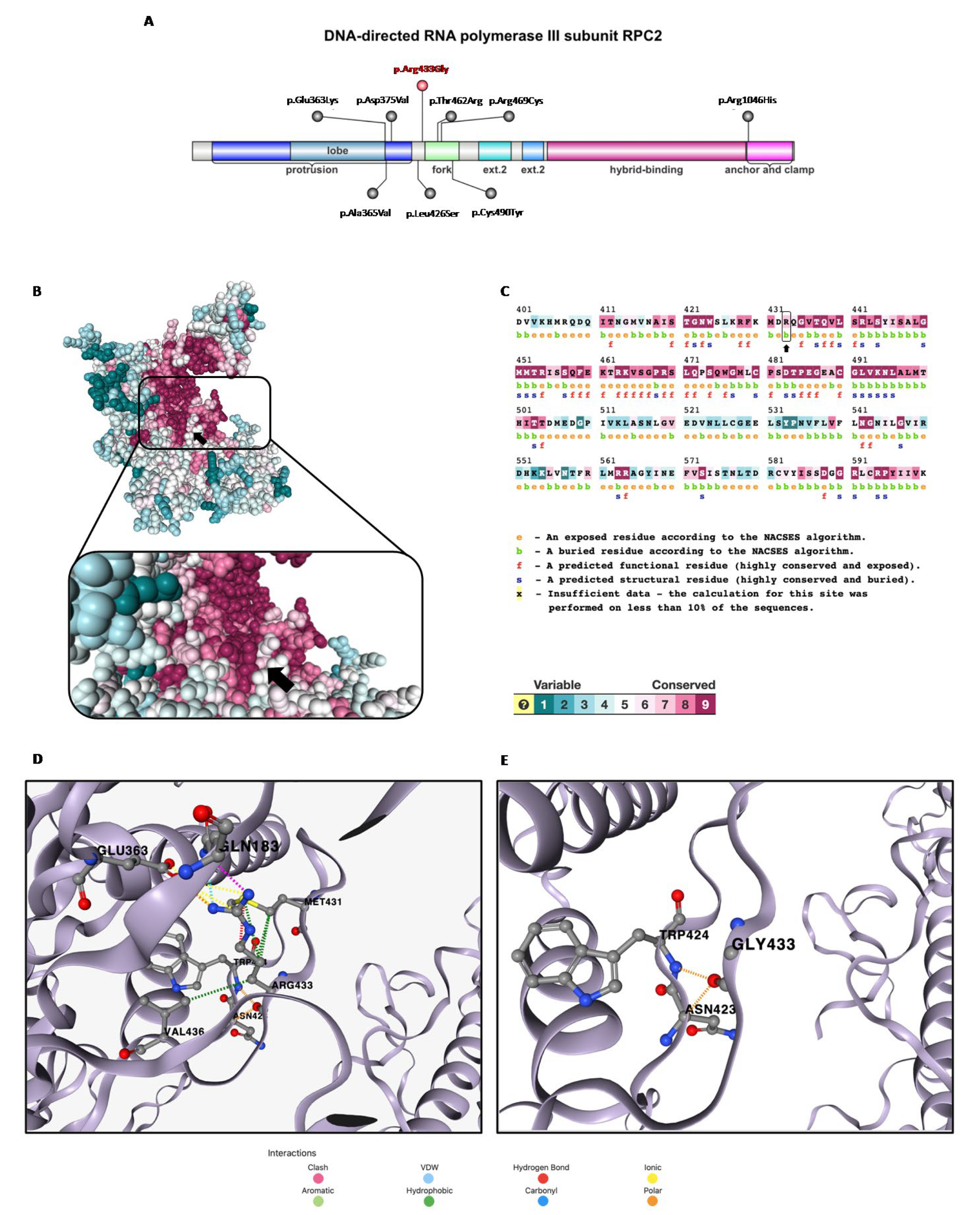
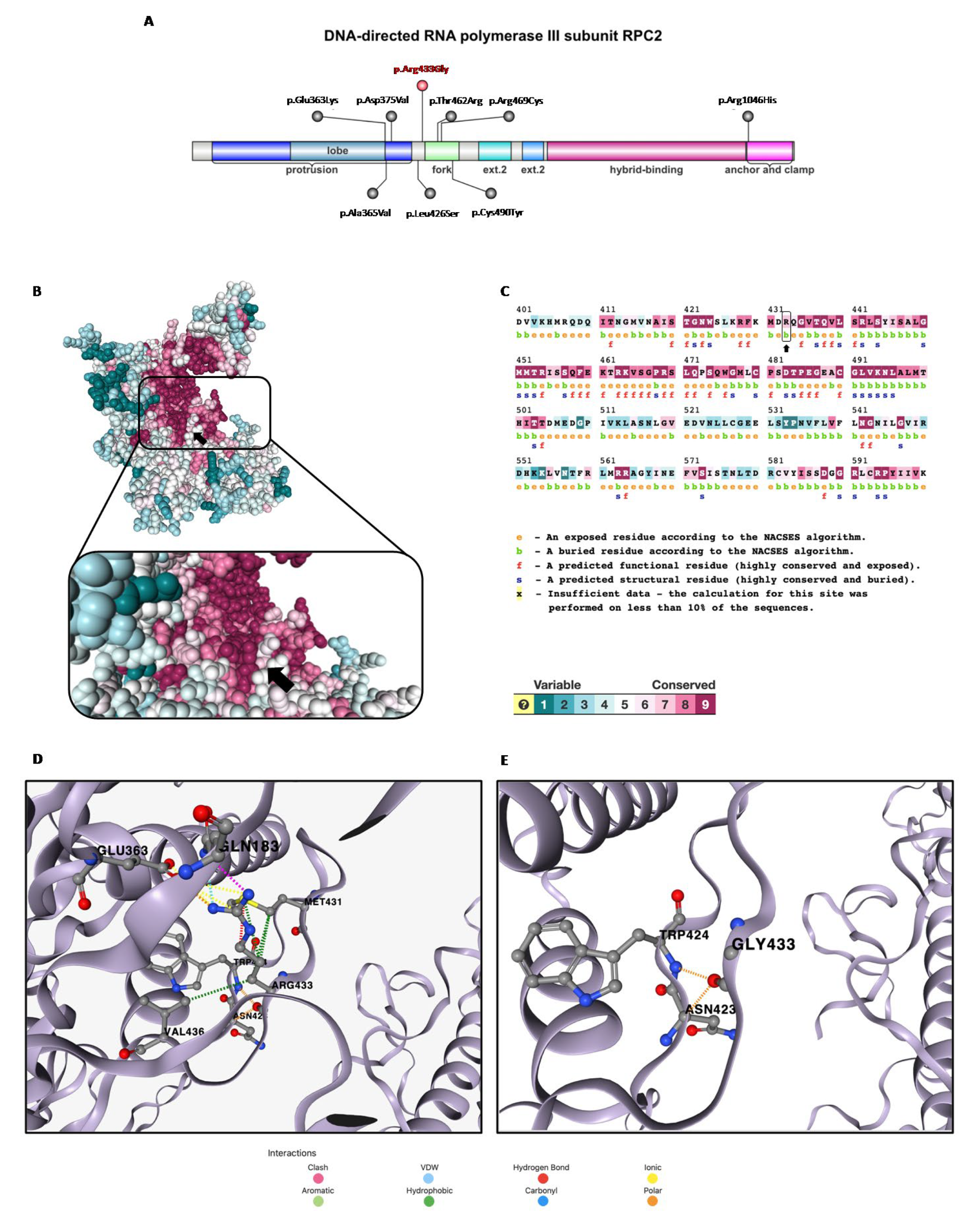
3.1. In Silico Analysis
3.2. Autosomal-Dominant POLR3B Phenotypic Spectrum
3.3. CES Secondary Molecular Findings
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dumay-Odelot, H.; Durrieu-Gaillard, S.; Da Silva, D.; Roeder, R.G.; Teichmann, M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle 2010, 9, 3687–3699. [Google Scholar] [CrossRef]
- Bernard, G.; Vanderver, A. POLR3-Related Leukodystrophy. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2012; pp. 1993–2023. [Google Scholar]
- Tétreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Harting, I.; Boltshauser, E.; Wiegand, G.; Koch, M.J.; Schmitt-Mechelke, T.; Martin, E.; Zschocke, J.; Uhlenberg, B.; Hoffmann, G.F.; et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 2005, 64, 1461–1464. [Google Scholar] [CrossRef]
- Wolf, N.I.; Harting, I.; Innes, A.M.; Patzer, S.; Zeitler, P.; Schneider, A.; Wolff, A.; Baier, K.; Zschocke, J.; Ebinger, F.; et al. Ataxia, delayed dentition and hypomyelination: A novel leukoencephalopathy. Neuropediatrics 2007, 38, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.Y.; Cheng, H.L.; Dong, H.L.; Yin, H.M.; Yuan, Y.; Meng, L.C.; Wu, Z.Y.; Yu, H. A de novo variant of POLR3B causes demyelinating Charcot-Marie-Tooth disease in a Chinese patient: A case report. BMC Neurol. 2021, 21, 402. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, D.; Pinard, M.; Gauthier, M.S.; Smith-Hicks, C.; Hoffman, T.L.; Wolf, N.I.; Oegema, R.; van Binsbergen, E.; Baskin, B.; Bernard, G.; et al. De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy. Am. J. Hum. Genet. 2021, 108, 186–193, Erratum in Am. J. Hum. Genet. 2022, 109, 759–763. [Google Scholar] [CrossRef]
- Ando, M.; Higuchi, Y.; Yuan, J.H.; Yoshimura, A.; Kitao, R.; Morimoto, T.; Taniguchi, T.; Takeuchi, M.; Takei, J.; Hiramatsu, Y.; et al. Novel de novo POLR3B mutations responsible for demyelinating Charcot-Marie-Tooth disease in Japan. Ann. Clin. Transl. Neurol. 2022, 9, 747–755. [Google Scholar] [CrossRef]
- Abascal-Palacios, G.; Ramsay, E.P.; Beuron, F.; Morris, E.; Vannini, A. Structural basis of RNA polymerase III transcription initiation. Nature 2018, 553, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Chorin, A.B.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Using evolutionary data to make sense of macromolecules with a “face-lifted” ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genomics 2015, 16 (Suppl. S8), S1. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Strupp, M.; Kremmyda, O.; Adamczyk, C.; Bottcher, N.; Muth, C.; Yip, C.W.; Bremova, T. Central ocular motor disorders, including gaze palsy and nystagmus. J. Neurol. 2014, 261 (Suppl. S2), S542–S558. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Li, D.; Zheng, Y.; Jiang, X. Case report: Biallelic variants in POLR3B gene lead to 4H leukodystrophy from the study of brother and sister. Medicine 2022, 101, e30350. [Google Scholar] [CrossRef] [PubMed]
- Jaques, C.S.; Escorcio-Bezerra, M.L.; Pedroso, J.L.; Barsottini, O.G.P. The Intersection Between Cerebellar Ataxia and Neuropa-thy: A Proposed Classification and a Diagnostic Approach. Cerebellum 2022, 21, 497–513. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colona, V.L.; Bertini, E.; Digilio, M.C.; D’Amico, A.; Novelli, A.; Pro, S.; Pisaneschi, E.; Nicita, F. A New Case of Autosomal-Dominant POLR3B-Related Disorder: Widening Genotypic and Phenotypic Spectrum. Brain Sci. 2023, 13, 1567. https://doi.org/10.3390/brainsci13111567
Colona VL, Bertini E, Digilio MC, D’Amico A, Novelli A, Pro S, Pisaneschi E, Nicita F. A New Case of Autosomal-Dominant POLR3B-Related Disorder: Widening Genotypic and Phenotypic Spectrum. Brain Sciences. 2023; 13(11):1567. https://doi.org/10.3390/brainsci13111567
Chicago/Turabian StyleColona, Vito Luigi, Enrico Bertini, Maria Cristina Digilio, Adele D’Amico, Antonio Novelli, Stefano Pro, Elisa Pisaneschi, and Francesco Nicita. 2023. "A New Case of Autosomal-Dominant POLR3B-Related Disorder: Widening Genotypic and Phenotypic Spectrum" Brain Sciences 13, no. 11: 1567. https://doi.org/10.3390/brainsci13111567
APA StyleColona, V. L., Bertini, E., Digilio, M. C., D’Amico, A., Novelli, A., Pro, S., Pisaneschi, E., & Nicita, F. (2023). A New Case of Autosomal-Dominant POLR3B-Related Disorder: Widening Genotypic and Phenotypic Spectrum. Brain Sciences, 13(11), 1567. https://doi.org/10.3390/brainsci13111567