
Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and Data Collection
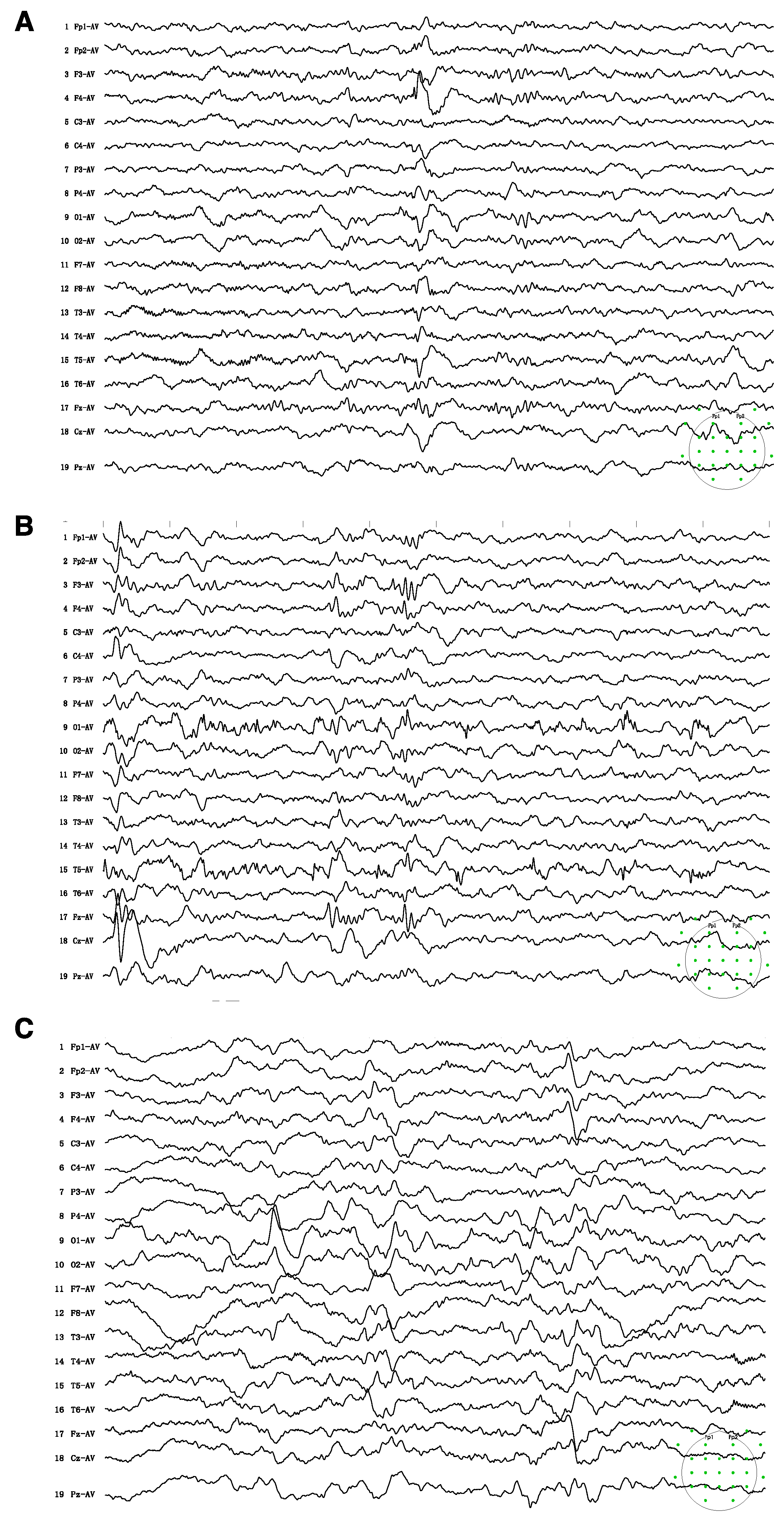
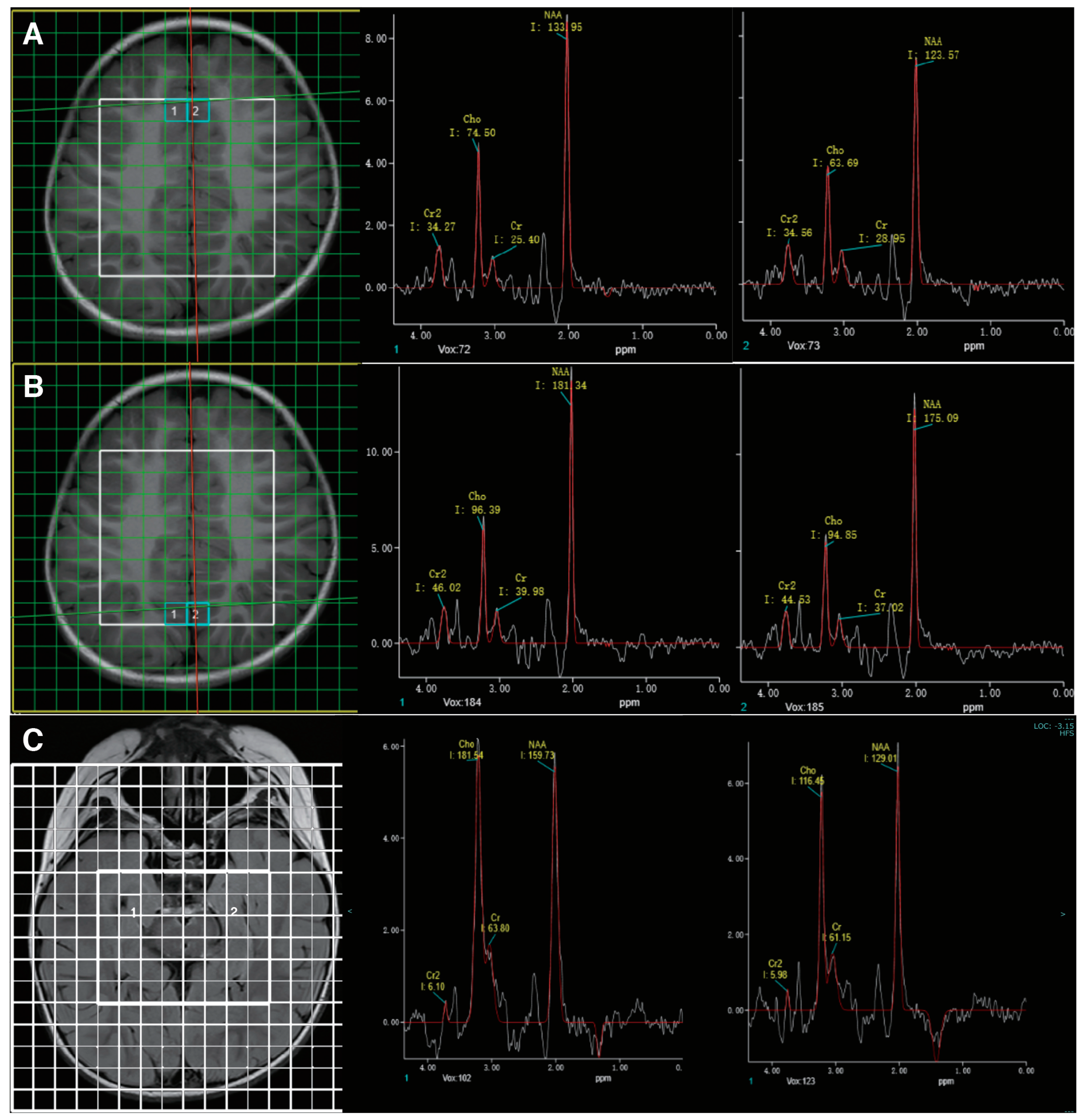
2.2. Biochemical and Image Analysis
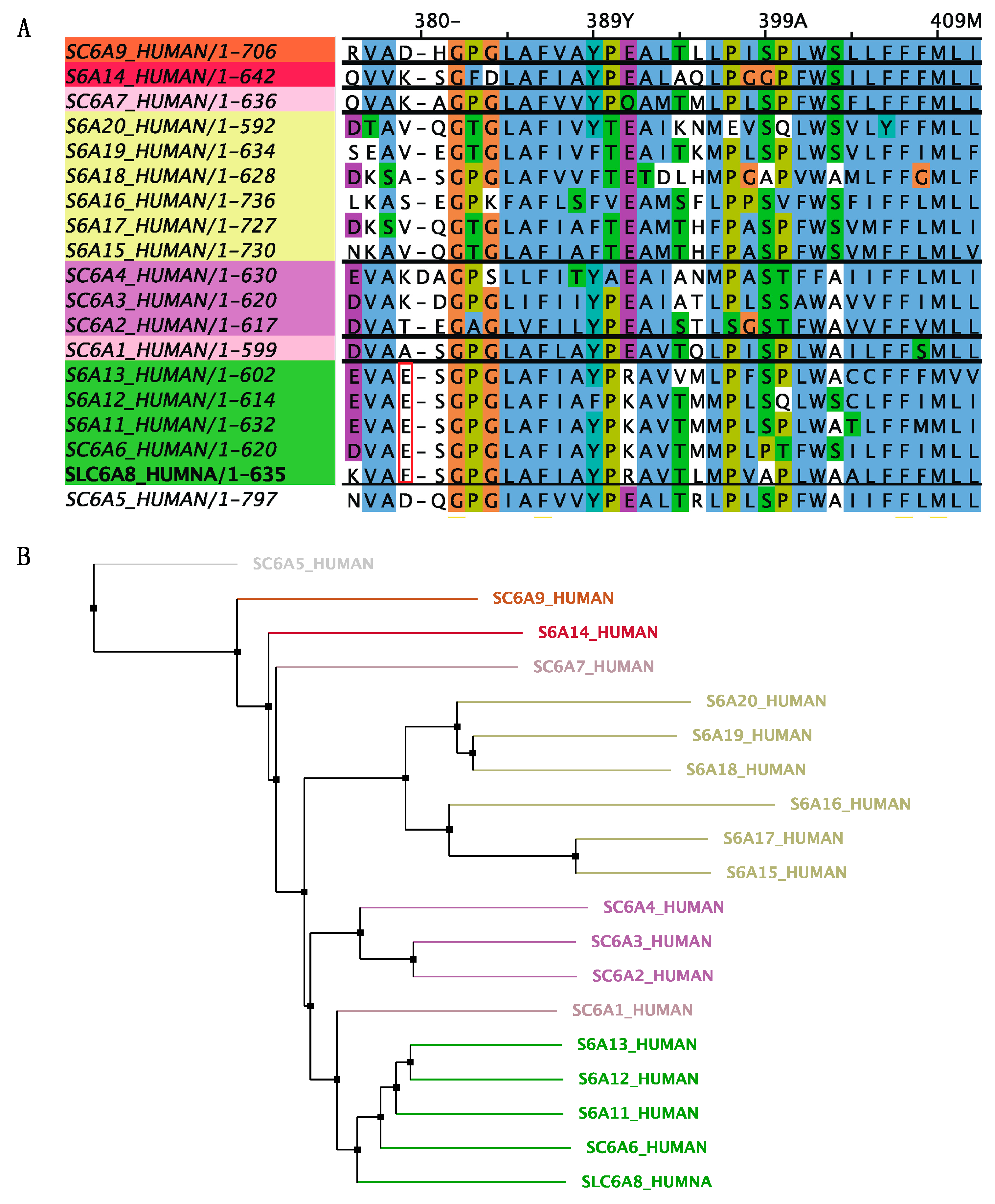
2.3. Genetic Sequencing and In Silico Analysis
2.4. Neuropsychological Assessment
2.5. Literature Search Method
3. Results
3.1. Case Presentation
3.2. In Silico Analysis of the Identified Variant
3.3. Treatment and Follow-Up
3.4. Systematic Literature Review of Creatine Treatment in CTD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braissant, O.; Henry, H.; Loup, M.; Eilers, B.; Bachmann, C. Endogenous synthesis and transport of creatine in the rat brain: An in situ hybridization study. Brain Res. Mol. Brain Res. 2001, 86, 193–201. [Google Scholar] [CrossRef]
- Exocytotic Release of Creatine in Rat Brain—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/16715490/ (accessed on 23 June 2023).
- Duran-Trio, L.; Fernandes-Pires, G.; Grosse, J.; Soro-Arnaiz, I.; Roux-Petronelli, C.; Binz, P.-A.; De Bock, K.; Cudalbu, C.; Sandi, C.; Braissant, O. Creatine transporter–deficient rat model shows motor dysfunction, cerebellar alterations, and muscle creatine deficiency without muscle atrophy. J. Inherit. Metab. Dis. 2022, 45, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.P.; Wyse, A.T.S. Creatine as a Neuroprotector: An Actor that Can Play Many Parts. Neurotox. Res. 2019, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, J.M.; Mancini, G.M.; Salomons, G.S. X-linked creatine transporter deficiency: Clinical aspects and pathophysiology. J. Inherit. Metab. Dis. 2014, 37, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; Degrauw, R.S.; Yntema, H.G.; Bahi, N.; Moraine, C.; Ropers, H.-H.; Fryns, J.-P.; Degrauw, T.J.; et al. High Prevalence of SLC6A8 Deficiency in X-Linked Mental Retardation. Am. J. Hum. Genet. 2004, 75, 97–105. [Google Scholar] [CrossRef]
- van de Kamp, J.M.; Betsalel, O.T.; Mercimek-Mahmutoglu, S.; Abulhoul, L.; Grünewald, S.; Anselm, I.; Azzouz, H.; Bratkovic, D.; de Brouwer, A.; Hamel, B.; et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J. Med. Genet. 2013, 50, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.S.; Verhoeven, N.M.; Roos, B.; Valongo, C.; Cardoso, M.L.; Vilarinho, L.; Salomons, G.S.; Jakobs, C. Creatine and guanidinoacetate: Diagnostic markers for inborn errors in creatine biosynthesis and transport. Mol. Genet. Metab. 2004, 82, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Screening for X-Linked Creatine Transporter (SLC6A8) Deficiency via Simultaneous Determination of Urinary Creatine to Creatinine Ratio by Tandem Mass-Spectrometry—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19188083/ (accessed on 12 September 2023).
- Rosenberg, E.H.; Muñoz, C.M.; Betsalel, O.T.; van Dooren, S.J.; Fernandez, M.; Jakobs, C.; Degrauw, T.J.; Kleefstra, T.; Schwartz, C.E.; Salomons, G.S. Functional characterization of missense variants in the creatine transporter gene (SLC6A8): Improved diagnostic application. Hum. Mutat. 2007, 28, 890–896. [Google Scholar] [CrossRef]
- Longo, N.; Ardon, O.; Vanzo, R.; Schwartz, E.; Pasquali, M. Disorders of creatine transport and metabolism. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 72–78. [Google Scholar] [CrossRef]
- Yu, H.; van Karnebeek, C.; Sinclair, G.; Hill, A.; Cui, H.; Zhang, V.W.; Wong, L.-J. Detection of a novel intragenic rearrangement in the creatine transporter gene by next generation sequencing. Mol. Genet. Metab. 2013, 110, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Kasai, Y.; Yokoyama, R.; Fujinawa, J.; Ganapathy, V.; Terasaki, T.; Hosoya, K.-I. The blood-brain barrier transport and cerebral distribution of guanidinoacetate in rats: Involvement of creatine and taurine transporters. J. Neurochem. 2009, 111, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Tachikawa, M.; Takanaga, H.; Shimizu, H.; Watanabe, M.; Hosoya, K.-I.; Terasaki, T. The Blood–Brain Barrier Creatine Transporter is a Major Pathway for Supplying Creatine to the Brain. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2002, 22, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Farr, C.V.; El-Kasaby, A.; Freissmuth, M.; Sucic, S. The Creatine Transporter Unfolded: A Knotty Premise in the Cerebral Creatine Deficiency Syndrome. Front. Synaptic Neurosci. 2020, 12, 588954. [Google Scholar] [CrossRef]
- van de Kamp, J.M.; Jakobs, C.; Gibson, K.M.; Salomons, G.S. New insights into creatine transporter deficiency: The importance of recycling creatine in the brain. J. Inherit. Metab. Dis. 2013, 36, 155–156. [Google Scholar] [CrossRef]
- Fons, C.; Sempere, A.; Arias, A.; López-Sala, A.; Póo, P.; Pineda, M.; Mas, A.; Vilaseca, M.A.; Salomons, G.S.; Ribes, A.; et al. Arginine Supplementation in Four Patients with X-Linked Creatine Transporter Defect. J. Inherit. Metab. Dis. 2008, 31, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Mercimek-Mahmutoglu, S.; Connolly, M.B.; Poskitt, K.J.; Horvath, G.A.; Lowry, N.; Salomons, G.S.; Casey, B.; Sin-clair, G.; Davis, C.; Jakobs, C.; et al. Treatment of Intractable Epilepsy in a Female with SLC6A8 Deficiency. Mol. Genet. Metab. 2010, 101, 409–412. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Boddaert, N.; Chabli, A.; Barbier, V.; Desguerre, I.; Philippe, A.; Afenjar, A.; Mazzuca, M.; Cheillan, D.; Munnich, A.; et al. Treatment by Oral Creatine, L-Arginine and L-Glycine in Six Severely Affected Patients with Creatine Transporter Defect. J. Inherit. Metab. Dis. 2012, 35, 151–157. [Google Scholar] [CrossRef]
- Chilosi, A.; Casarano, M.; Comparini, A.; Battaglia, F.M.; Mancardi, M.M.; Schiaffino, C.; Tosetti, M.; Leuzzi, V.; Battini, R.; Cioni, G. Neuropsychological profile and clinical effects of arginine treatment in children with creatine transport deficiency. Orphanet J. Rare Dis. 2012, 7, 43. [Google Scholar] [CrossRef]
- van de Kamp, J.M.; Pouwels, P.J.W.; Aarsen, F.K.; ten Hoopen, L.W.; Knol, D.L.; de Klerk, J.B.; de Coo, I.F.; Huijmans, J.G.M.; Jakobs, C.; van der Knaap, M.S.; et al. Long-Term Follow-up and Treatment in Nine Boys with X-Linked Creatine Transporter Defect. J. Inherit. Metab. Dis. 2012, 35, 141. [Google Scholar] [CrossRef]
- Dunbar, M.; Jaggumantri, S.; Sargent, M.; Stockler-Ipsiroglu, S.; van Karnebeek, C.D.M. Treatment of X-Linked Creatine Transporter (SLC6A8) Deficiency: Systematic Review of the Literature and Three New Cases. Mol. Genet. Metab. 2014, 112, 259–274. [Google Scholar] [CrossRef]
- Jaggumantri, S.; Dunbar, M.; Edgar, V.; Mignone, C.; Newlove, T.; Elango, R.; Collet, J.P.; Sargent, M.; Stockler-Ipsiroglu, S.; van Karnebeek, C.D.M. Treatment of Creatine Transporter (SLC6A8) Deficiency With Oral S-Adenosyl Methionine as Adjunct to L-Arginine, Glycine, and Creatine Supplements. Pediatr. Neurol. 2015, 53, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Bruun, T.U.J.; Sidky, S.; Bandeira, A.O.; Debray, F.-G.; Ficicioglu, C.; Goldstein, J.; Joost, K.; Koeberl, D.D.; Luísa, D.; Nassogne, M.-C.; et al. Treatment outcome of creatine transporter deficiency: International retrospective cohort study. Metab. Brain Dis. 2018, 33, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Jangid, N.; Surana, P.; Salmonos, G.; Jain, V. Creatine Transporter Deficiency, an Underdiagnosed Cause of Male Intellectual Disability. BMJ Case Rep. 2020, 13, e237542. [Google Scholar] [CrossRef] [PubMed]
- Yıldız, Y.; Göçmen, R.; Yaramış, A.; Coşkun, T.; Haliloğlu, G. Creatine Transporter Deficiency Presenting as Au-tism Spectrum Disorder. Pediatrics 2020, 146, e20193460. [Google Scholar] [CrossRef]
- Brugger, M.; Brunet, T.; Wagner, M.; Orec, L.E.; Schwaibold, E.M.C.; Boy, N. Locus Heterogeneity in Two Siblings Presenting with Developmental Delay, Intellectual Disability and Autism Spectrum Disorder. Gene 2021, 768, 145260. [Google Scholar] [CrossRef]
- Sun, W.; Wang, Y.; Wu, M.; Wu, H.; Peng, X.; Shi, Y.; Xiao, F.; Wu, B.; Zhou, W.; Lu, W. Fourteen Cases of Cere-bral Creatine Deficiency Syndrome in Children: A Cohort Study in China. Transl. Pediatr. 2023, 12, 927–937. [Google Scholar] [CrossRef]
- Bizzi, A.; Bugiani, M.; Salomons, G.S.; Hunneman, D.H.; Moroni, I.; Estienne, M.; Danesi, U.; Jakobs, C.; Uziel, G. X-linked creatine deficiency syndrome: A novel mutation in creatine transporter geneSLC6A8. Ann. Neurol. 2002, 52, 227–231. [Google Scholar] [CrossRef]
- Degrauw, T.J.; Salomons, G.S.; Cecil, K.M.; Chuck, G.; Newmeyer, A.; Schapiro, M.B.; Jakobs, C. Congenital Creatine Transporter Deficiency. Neuropediatrics 2002, 33, 232–238. [Google Scholar] [CrossRef]
- Shi, K.; Zhao, H.; Xu, S.; Han, H.; Li, W. Treatment efficacy of high-dose creatine supplementation in a child with creatine transporter (SLC6A8) deficiency. Mol. Genet. Genom. Med. 2021, 9, e1640. [Google Scholar] [CrossRef]
- Forbes, S.C.; Cordingley, D.M.; Cornish, S.M.; Gualano, B.; Roschel, H.; Ostojic, S.M.; Rawson, E.S.; Roy, B.D.; Prokopidis, K.; Giannos, P.; et al. Effects of Creatine Supplementation on Brain Function and Health. Nutrients 2022, 14, 921. [Google Scholar] [CrossRef]
- Fons, C.; Campistol, J. Creatine Defects and Central Nervous System. Semin. Pediatr. Neurol. 2016, 23, 285–289. [Google Scholar] [CrossRef]
- Allen, P.J. Creatine metabolism and psychiatric disorders: Does creatine supplementation have therapeutic value? Neurosci. Biobehav. Rev. 2012, 36, 1442–1462. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Yücel, M.; Pantelis, C.; Berk, M. Neurobiology of Schizophrenia Spectrum Disorders: The Role of Oxidative Stress. Ann. Acad. Med. Singap. 2009, 38, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.; Renshaw, P.F. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 2005, 10, 900–919. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.I.; Ressler, K.J.; Binder, E.; Nemeroff, C.B. The Neurobiology of Anxiety Disorders: Brain Imaging, Genetics, and Psychoneuroendocrinology. Psychiatr. Clin. N. Am. 2009, 32, 549–575. [Google Scholar] [CrossRef]
- Alraddadi, E.A.; Khojah, A.M.; Alamri, F.F.; Kecheck, H.K.; Altaf, W.F.; Khouqeer, Y. Potential role of creatine as an anticonvulsant agent: Evidence from preclinical studies. Front. Neurosci. 2023, 17, 1201971. [Google Scholar] [CrossRef]
- Royes, L.F.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S.; da Silva, L.G.M.; Malfatti, C.R.M.; Schneider, P.H.; Braga, A.L.; Wajner, M.; Mello, C.F. Creatine protects against the convulsive behavior and lactate production elicited by the intrastriatal injection of methylmalonate. Neuroscience 2003, 118, 1079–1090. [Google Scholar] [CrossRef]
- Royes, L.F.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S.; Myskiw, J.d.C.; Fiorenza, N.G.; Petry, J.C.; Coelho, R.C.; Mello, C.F. Effectiveness of creatine monohydrate on seizures and oxidative damage induced by methylmalonate. Pharmacol. Biochem. Behav. 2006, 83, 136–144. [Google Scholar] [CrossRef]
- Magni, D.V.; Oliveira, M.S.; Furian, A.F.; Fiorenza, N.G.; Fighera, M.R.; Ferreira, J.; Mello, C.F.; Royes, L.F.F. Creatine decreases convulsions and neurochemical alterations induced by glutaric acid in rats. Brain Res. 2007, 1185, 336–345. [Google Scholar] [CrossRef]
- Salomons, G.S.; van Dooren, S.J.; Verhoeven, N.M.; Cecil, K.M.; Ball, W.S.; Degrauw, T.J.; Jakobs, C. X-Linked Creatine-Transporter Gene (SLC6A8) Defect: A New Creatine-Deficiency Syndrome. Am. J. Hum. Genet. 2001, 68, 1497–1500. [Google Scholar] [CrossRef]
- Osaka, H.; Takagi, A.; Tsuyusaki, Y.; Wada, T.; Iai, M.; Yamashita, S.; Shimbo, H.; Saitsu, H.; Salomons, G.S.; Jakobs, C.; et al. Contiguous deletion of SLC6A8 and BAP31 in a patient with severe dystonia and sensorineural deafness. Mol. Genet. Metab. 2012, 106, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Banci, G.; Martini, R.; Ecker, G.F. Studies of structural determinants of substrate binding in the Creatine Transporter (CreaT, SLC6A8) using molecular models. Sci. Rep. 2020, 10, 6241. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.D.; Zelt, N.B.; Saldivar, R.; Kuntz, C.P.; Chen, S.; Penn, W.D.; Bonneau, R.; Leman, J.K.; Schlebach, J.P. Classification of the Molecular Defects Associated with Pathogenic Variants of the SLC6A8 Creatine Transporter. Biochemistry 2020, 59, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.L. Functional Insights into the Creatine Transporter. In Creatine and Creatine Kinase in Health and Disease; Salomons, G.S., Wyss, M., Eds.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2007; ISBN 978-1-4020-6486-9. [Google Scholar]
- Betsalel, O.T.; Pop, A.; Rosenberg, E.H.; Fernandez-Ojeda, M.; Creatine Transporter Research Group; Jakobs, C.; Salomons, G.S. Detection of variants in SLC6A8 and functional analysis of unclassified missense variants. Mol. Genet. Metab. 2012, 105, 596–601. [Google Scholar] [CrossRef]
- Clark, A.J.; Rosenberg, E.H.; Almeida, L.S.; Wood, T.C.; Jakobs, C.; Stevenson, R.E.; Schwartz, C.E.; Salomons, G.S. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum. Genet. 2006, 119, 604–610. [Google Scholar] [CrossRef]
- Braissant, O. Creatine and guanidinoacetate transport at blood-brain and blood-cerebrospinal fluid barriers. J. Inherit. Metab. Dis. 2012, 35, 655–664. [Google Scholar] [CrossRef]
- Braissant, O.; Henry, H.; Béard, E.; Uldry, J. Creatine deficiency syndromes and the importance of creatine synthesis in the brain. Amino Acids 2011, 40, 1315–1324. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. Creatine: Endogenous Metabolite, Dietary, and Therapeutic Supplement. Available online: https://www.annualreviews.org/doi/abs/10.1146/annurev.nutr.27.061406.093621 (accessed on 17 September 2023).
- Ullio-Gamboa, G.; Udobi, K.C.; Dezard, S.; Perna, M.K.; Miles, K.N.; Costa, N.; Taran, F.; Pruvost, A.; Benoit, J.-P.; Skelton, M.R.; et al. Dodecyl creatine ester-loaded nanoemulsion as a promising therapy for creatine transporter deficiency. Nanomedicine 2019, 14, 1579–1593. [Google Scholar] [CrossRef]
- Schjelderup, J.; Hope, S.; Vatshelle, C.; van Karnebeek, C.D.M. Treatment experience in two adults with creatinfe transporter deficiency. Mol. Genet. Metab. Rep. 2021, 27, 100731. [Google Scholar] [CrossRef]
- El-Kasaby, A.; Kasture, A.; Koban, F.; Hotka, M.; Asjad, H.M.M.; Kubista, H.; Freissmuth, M.; Sucic, S. Rescue by 4-phenylbutyrate of several misfolded creatine transporter-1 variants linked to the creatine transporter deficiency syndrome. Neuropharmacology 2019, 161, 107572. [Google Scholar] [CrossRef]
- Cacciante, F.; Gennaro, M.; Sagona, G.; Mazziotti, R.; Lupori, L.; Cerri, E.; Putignano, E.; Butt, M.; Do, M.-H.T.; McKew, J.C.; et al. Cyclocreatine treatment ameliorates the cognitive, autistic and epileptic phenotype in a mouse model of Creatine Transporter Deficiency. Sci. Rep. 2020, 10, 18361. [Google Scholar] [CrossRef] [PubMed]
- In Vivo Neuroprotection by a Creatine-Derived Compound: Phosphocreatine–Mg-Complex Acetate—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0006899309011743 (accessed on 26 July 2023).
- Burov, S.; Leko, M.; Dorosh, M.; Dobrodumov, A.; Veselkina, O. Creatinyl amino acids-new hybrid compounds with neuroprotective activity. J. Pept. Sci. 2011, 17, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, Y.; DeGrauw, T.J.; Lindquist, D.M.; Blanco, V.M.; Pyne-Geithman, G.J.; Daikoku, T.; Chambers, J.B.; Benoit, S.C.; Clark, J.F. Cyclocreatine treatment improves cognition in mice with creatine transporter deficiency. J. Clin. Investig. 2012, 122, 2837–2846. [Google Scholar] [CrossRef]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Chtarto, A.; Bockstael, O.; Gebara, E.; Vermoesen, K.; Melas, C.; Pythoud, C.; Levivier, M.; De Witte, O.; Luthi-Carter, R.; Clinkers, R.; et al. An Adeno-Associated Virus-Based Intracellular Sensor of Pathological Nuclear Factor-κB Activation for Disease-Inducible Gene Transfer. PLoS ONE 2013, 8, e53156. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Subjects | Treatment | Outcome | |
|---|---|---|---|---|
| N/Sex | Age | |||
| Fons et al., 2008 [17] | 4/M | 9–16 | Arg 400 mg/kg/d for 9 mo | No improvement in neuropsychological assessment; cerebral Cr not increased |
| Mercimek-Mahmutoglu et al., 2010 [18] | 1/F | 6 | Cr 300 mg/kg/d, Arg 450 mg/kg/d + Gly 150 mg/kg/d for 28 mo | Resolution of intractable seizures; no other clinical effect; marginally increased cerebral Cr |
| Valayannopoulos et al., 2011 [19] | 4/M, 2/F | 2–16 | Cr 400 mg/kg/d for 6 mo, then Cr + Arg 200 mg/kg/d + Gly 200 mg/kg/d for 12 mo, then Arg 200 mg/kg/d + Gly 200 mg/kg/d for 24 mo | No cognitive or psychiatric improvement, improvement in gross motor function in two males; cerebral Cr not increased |
| Chilosi et al., 2012 [20] | 3/M | 5–8.5 | Arg 300 mg/kg/d for 24–36 mo | Seizure control; improvement in language and adaptive skills; creatine peak enhancement |
| van de Kamp et al., 2012 [21] | 9/M | 0–10 | Cr 400 mg/kg/d + Arg 400 mg/kg/d + Gly 150 mg/kg/d for 48–72 mo | No measurable clinical improvement or deuteriation; cerebral Cr not increased |
| Dunbar et al., 2014 [22] | 3/M | 3–4 | Cr 400 mg/kg/d, Arg 400 mg/kg/d + Gly 150 mg/kg/d for 28 mo for 5–33 mo | Improvement in neuropsychic functioning and speech; increased Cr/c by 1.2–4.3 times |
| Jaggumantri et al., 2015 * [23] | 1/M | 8 | SAM (50 mg/kg/d) + Arg, Gly, and Cr for 3 mo | Significant improvement in speech/language skills; cerebral Cr not increased on MRS |
| Bruun et al., 2018 [24] | 14/M, 3/F | 1.2–7 | Various regimens of combined Cr, Arg, and Gly therapy | None of the males showed either deterioration or improvements; two females showed improvements in the clinical severity score |
| Jangid et al., 2020 [25] | 1/M | 3 | Cr 400 mg/kg/day + Gly 150 mg/kg/day for 12 mo, Arg 150 mg/kg/day for 3 mo | Modest motor and cognitive improvement |
| Yıldız et al., 2020 [26] | 1/M | 6 | Cr 100 mg/kg/d, Arg 400 mg/kg/d, Gly 150 mg/kg/d, and SAM (up to 50 mg/kg/d) for 24 mo | Mild, subjective improvement in attention, expressive language, and behavior |
| Brugger et al., 2021 [27] | 1/F | 8.6 | Gly 200 mg/kg/d, Arg 400 mg/kg/d, and Cr 400 mg/kg/d for 18 mo | Improvement in fine motor function, social behavior, and weight gain |
| Sun et al., 2023 [28] | 2/M | 0.7–2 | Cr 400 mg/kg/d, Arg 300 mg/kg/d, Gly 150 mg/kg/d + Cr gluconate 400 mg/kg/d for 2–3 mo | Improvement in motor skills and cognition |
| this study | 1/M | 0.75 | Cr 400 mg/kg/d + Gly 150 mg/kg/d for 6 mo | Improvement in cognitive function, speech skills, and social interaction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xu, S. Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review. Brain Sci. 2023, 13, 1382. https://doi.org/10.3390/brainsci13101382
Li J, Xu S. Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review. Brain Sciences. 2023; 13(10):1382. https://doi.org/10.3390/brainsci13101382
Chicago/Turabian StyleLi, Jiaqing, and Sanqing Xu. 2023. "Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review" Brain Sciences 13, no. 10: 1382. https://doi.org/10.3390/brainsci13101382
APA StyleLi, J., & Xu, S. (2023). Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review. Brain Sciences, 13(10), 1382. https://doi.org/10.3390/brainsci13101382