
Young Adults with a Parent with Dementia Show Early Abnormalities in Brain Activity and Brain Volume in the Hippocampus: A Matched Case-Control Study

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Measures of Cognition
2.3. MRI Acquisition and Preprocessing
2.4. Statistical Analyses
3. Results
3.1. Sample Characteristics
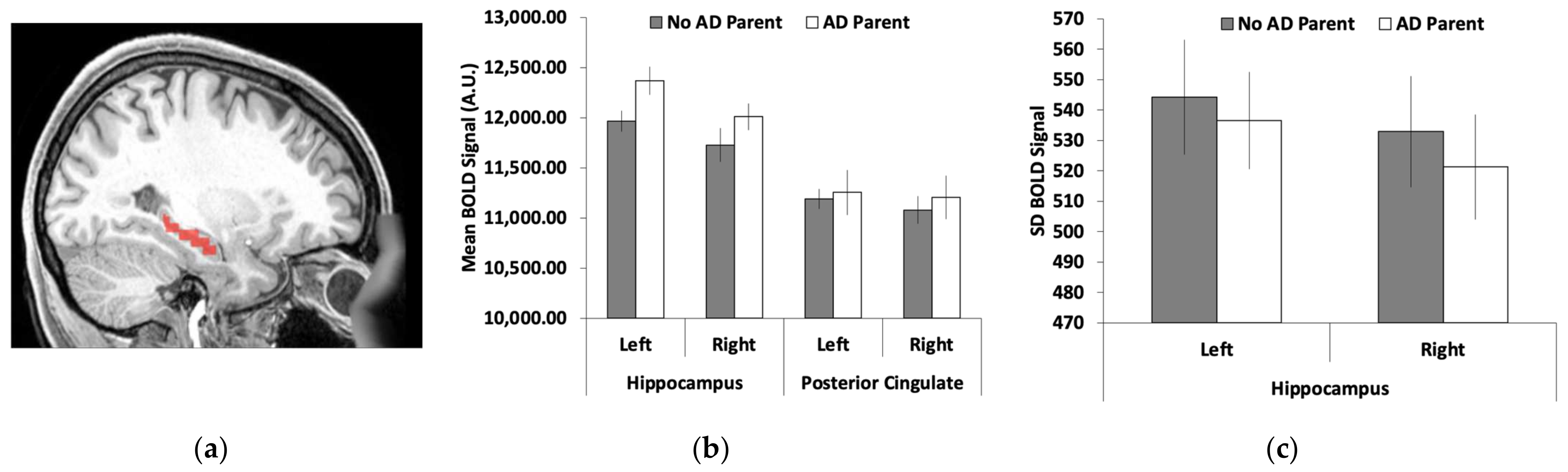
3.2. Hippocamal Brain Activity
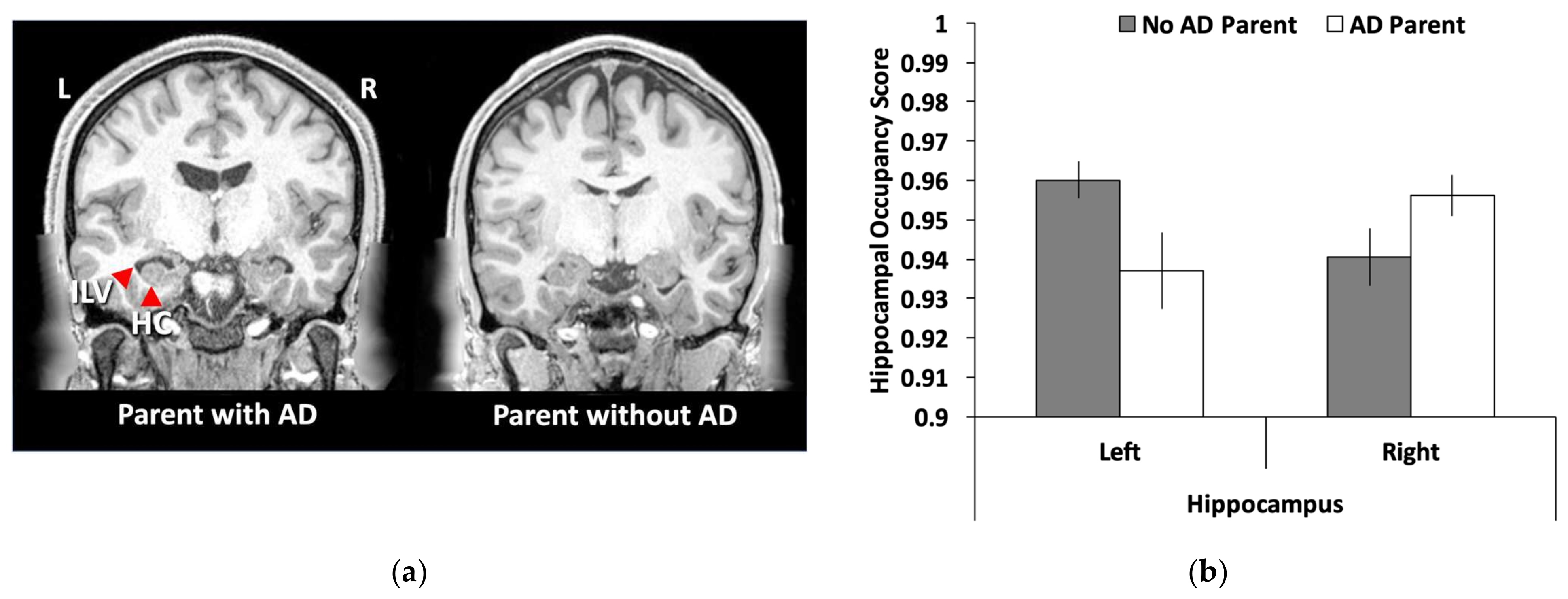
3.3. Hippocampal Volume
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, H.; Seo, S.W.; Kim, J.H.; Kim, C.; Ye, B.S.; Kim, G.H.; Noh, Y.; Kim, H.J.; Yoon, C.W.; Seong, J.K.; et al. Changes in subcortical structures in early- versus late-onset Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1740–1747. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R., Jr.; Aisen, P.S. Testing the right target and right drug at the right stage. Sci Transl. Med. 2011, 3, 111cm33. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Ohm, T.G.; Müller, H.; Braak, H.; Bohl, J. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer’s disease-related neurofibrillary changes. Neuroscience 1995, 64, 209–217. [Google Scholar] [CrossRef]
- Braak, E.; Griffing, K.; Arai, K.; Bohl, J.; Bratzke, H.; Braak, H. Neuropathology of Alzheimer’s disease: What is new since A. Alzheimer? Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Swaminathan, S.; Shen, L.; Foroud, T.M.; Hakonarson, H.; Huentelman, M.J.; Aisen, P.S.; et al. APOE and BCHE as modulators of cerebral amyloid deposition: A florbetapir PET genome-wide association study. Mol. Psychiatry 2014, 19, 351–357. [Google Scholar] [CrossRef]
- Donix, M.; Burggren, A.C.; Suthana, N.A.; Siddarth, P.; Ekstrom, A.D.; Krupa, A.K.; Jones, M.; Martin-Harris, L.; Ercoli, L.M.; Miller, K.J.; et al. Family history of Alzheimer’s disease and hippocampal structure in healthy people. Am. J. Psychiatry 2010, 167, 1399–1406. [Google Scholar] [CrossRef]
- Schuff, N.; Woerner, N.; Boreta, L.; Kornfield, T.; Shaw, L.M.; Trojanowski, J.Q.; Thompson, P.M.; Jack, C.R., Jr.; Weiner, M.W. Alzheimer’s; Disease Neuroimaging Initiative MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain 2009, 132, 1067–1077. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Vemuri, P.; Weigand, S.D.; Senjem, M.L.; Zeng, G.; Bernstein, M.A.; Gunter, J.L.; Pankratz, V.S.; Aisen, P.S.; et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain 2010, 133, 3336–3348. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, K.M.; Kennedy, K.M.; Devous, M.D.; Rieck, J.R.; Hebrank, A.C.; Diaz-Arrastia, R.; Mathews, D.; Park, D.C. Amyloid burden in healthy aging: Regional distribution and cognitive consequences. Neurology 2012, 78, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Dowell, N.G.; Evans, S.L.; Tofts, P.S.; King, S.L.; Tabet, N.; Rusted, J.M. Structural and resting-state MRI detects regional brain differences in young and mid-age healthy APOE-e4 carriers compared with non-APOE-e4 carriers. NMR Biomed. 2016, 29, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Filippini, N.; MacIntosh, B.J.; Hough, M.G.; Goodwin, G.M.; Frisoni, G.B.; Smith, S.M.; Matthews, P.M.; Beckmann, C.F.; Mackay, C.E. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. USA 2009, 106, 7209–7214. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, O.C.; Xu, G.; Dowling, N.M.; Bendlin, B.B.; Larue, A.; Hermann, B.P.; Koscik, R.; Jonaitis, E.; Rowley, H.A.; Carlsson, C.M.; et al. Family history of Alzheimer disease predicts hippocampal atrophy in healthy middle-aged adults. Neurology 2012, 78, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Kucikova, L.; Goerdten, J.; Dounavi, M.E.; Mak, E.; Su, L.; Waldman, A.D.; Danso, S.; Muniz-Terrera, G.; Ritchie, C.W. Resting-state brain connectivity in healthy young and middle-aged adults at risk of progressive Alzheimer’s disease. Neurosci. Biobehav. Rev. 2021, 129, 142–153. [Google Scholar] [CrossRef]
- McDonough, I.M.; Festini, S.B.; Wood, M.M. Risk for Alzheimer’s disease: A review of long-term episodic memory encoding and retrieval fMRI studies. Ageing Res. Rev. 2020, 62, 101133. [Google Scholar] [CrossRef]
- Martinez, M.; Campion, D.; Brice, A.; Hannequin, D.; Dubois, B.; Didierjean, O.; Michon, A.; Thomas-Anterion, C.; Puel, M.; Frebourg, T.; et al. Apolipoprotein E ϵ4 allele and familial aggregation of Alzheimer disease. Arch. Neurol. 1998, 55, 810–816. [Google Scholar] [CrossRef]
- Green, R.C.; Cupples, L.A.; Go, R.; Benke, K.S.; Edeki, T.; Griffith, P.A.; Williams, M.; Hipps, Y.; Graff-Radford, N.; Bachman, D.; et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002, 287, 329–336. [Google Scholar] [CrossRef]
- Cupples, L.A.; Farrer, L.A.; Sadovnick, A.D.; Relkin, N.; Whitehouse, P.; Green, R.C. Estimating risk curves for first-degree relatives of patients with Alzheimer’s disease: The REVEAL study. Genet. Med. 2004, 6, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.S.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele œµ4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Slooter, A.J.; Cruts, M.; Kalmijn, S.; Hofman, A.; Breteler, M.M.; Van Broeckhoven, C.; van Duijn, C.M. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch. Neurol. 1998, 55, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Berti, V.; Mosconi, L.; Glodzik, L.; Li, Y.; Murray, J.; De Santi, S.; Pupi, A.; Tsui, W.; De Leon, M.J. Structural brain changes in normal individuals with a maternal history of Alzheimer’s. Neurobiol. Aging 2011, 32, 2325.e17–2325.e26. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.S.; Yousem, D.M.; Cristinzio, C.; Kusevic, I.; Yassa, M.A.; Caffo, B.S.; Zeger, S.L. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain 2006, 129, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Roe, C.M.; Snyder, A.Z.; Brier, M.R.; Thomas, J.B.; Xiong, C.; Benzinger, T.L.; Morris, J.C.; Ances, B.M. Alzheimer disease family history impacts resting state functional connectivity. Ann. Neurol. 2012, 72, 571–577. [Google Scholar] [CrossRef] [PubMed]
- McDonough, I.M.; Allen, R.S. Biological markers of aging and mental health: A seed and soil model of neurocognitive disorders. Aging Ment. Health 2019, 23, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Buckner, R.L.; Sepulcre, J.; Talukdar, T.; Krienen, F.M.; Liu, H.; Hedden, T.; Andrews-Hanna, J.R.; Sperling, R.A.; Johnson, K.A. Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci. 2009, 29, 1860–1873. [Google Scholar] [CrossRef]
- Sperling, R.A.; Dickerson, B.C.; Pihlajamaki, M.; Vannini, P.; LaViolette, P.S.; Vitolo, O.V.; Hedden, T.; Becker, J.A.; Rentz, D.M.; Selkoe, D.J.; et al. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010, 12, 27–43. [Google Scholar] [CrossRef]
- Greicius, M.D.; Srivastava, G.; Reiss, A.L.; Menon, V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc. Natl. Acad. Sci. USA 2004, 101, 4637–4642. [Google Scholar] [CrossRef]
- Vincent, J.L.; Snyder, A.Z.; Fox, M.D.; Shannon, B.J.; Andrews, J.R.; Raichle, M.E.; Buckner, R.L. Coherent spontaneous activity identifies a hippocampal-parietal memory network. J. Neurophysiol. 2006, 96, 3517–3531. [Google Scholar] [CrossRef]
- Westlye, E.T.; Lundervold, A.; Rootwelt, H.; Lundervold, A.J.; Westlye, L.T. Increased hippocampal default mode synchronization during rest in middle-aged and elderly APOE Œµ4 carriers: Relationships with memory performance. J. Neurosci. 2011, 31, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Fenstermacher, E.; Bates, J.; Blacker, D.; Sperling, R.A.; Dickerson, B.C. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J. Neurol. Neurosurg. Psychiatry 2008, 79, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Sperling, R.A. Functional abnormalities of the medial temporal lobe memory system in mild cognitive impairment and Alzheimer’s disease: Insights from functional MRI studies. Neuropsychologia 2008, 46, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Park, D.C.; McDonough, I.M. The dynamic aging mind: Revelations from functional neuroimaging research. Perspect. Psychol. Sci. 2013, 8, 62–67. [Google Scholar] [CrossRef]
- Stern, E.A.; Bacskai, B.J.; Hickey, G.A.; Attenello, F.J.; Lombardo, J.A.; Hyman, B.T. Cortical synaptic integration in vivo is disrupted by amyloid-Œ≤ plaques. J. Neurosci. 2004, 24, 4535–4540. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef]
- Small, S.A. Isolating pathogenic mechanisms embedded within the hippocampal circuit through regional vulnerability. Neuron 2014, 84, 32–39. [Google Scholar] [CrossRef]
- Wig, G.S.; Grafton, S.T.; Demos, K.E.; Wolford, G.L.; Petersen, S.E.; Kelley, W.M. Medial temporal lobe BOLD activity at rest predicts individual differences in memory ability in healthy young adults. Proc. Natl. Acad. Sci. USA 2008, 105, 18555–18560. [Google Scholar] [CrossRef]
- Grady, C. The cognitive neuroscience of ageing. Nat. Rev. Neurosci. 2012, 13, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Garrett, D.D.; Kovacevic, N.; McIntosh, A.R.; Grady, C.L. Blood oxygen level-dependent signal variability is more than just noise. J. Neurosci. 2010, 30, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Samanez-Larkin, G.R.; Kuhnen, C.M.; Yoo, D.J.; Knutson, B. Variability in nucleus accumbens activity mediates age-related suboptimal financial risk taking. J. Neurosci. 2010, 30, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.C.; Smith, S.M.; Barch, D.M.; Behrens, T.E.; Yacoub, E.; Ugurbil, K.; for the Wu-Minn HCP Consortium. The WU-Minn human connectome project: An overview. Neuroimage 2013, 80, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M.; Burgess, G.C.; Harms, M.P.; Petersen, S.E.; Schlaggar, B.L.; Corbetta, M.; Glasser, M.F.; Curtiss, S.; Dixit, S.; Feldt, C.; et al. Function in the human connectome: Task-fMRI and individual differences in behavior. Neuroimage 2013, 80, 169–189. [Google Scholar] [CrossRef]
- Gur, R.C.; Richard, J.; Hughett, P.; Calkins, M.E.; Macy, L.; Bilker, W.B.; Brensinger, C.; Gur, R.E. A cognitive neuroscience-based computerized battery for efficient measurement of individual differences: Standardization and initial construct validation. J. Neurosci. Methods 2010, 187, 254–262. [Google Scholar] [CrossRef]
- Weintraub, S.; Dikmen, S.S.; Heaton, R.K.; Tulsky, D.S.; Zelazo, P.D.; Bauer, P.J.; Carlozzi, N.E.; Slotkin, J.; Blitz, D.; Wallner-Allen, K.; et al. Cognition assessment using the NIH Toolbox. Neurology 2013, 80, S54–S64. [Google Scholar] [CrossRef]
- Gershon, R.C.; Slotkin, J.; Manly, J.J.; Blitz, D.L.; Beaumont, J.L.; Schnipke, D.; Wallner-Allen, K.; Golinkoff, R.M.; Gleason, J.B.; Hirsh-Pasek, K.; et al. IV. NIH Toolbox Cognition Battery (CB): Measuring language (vocabulary comprehension and reading decoding). Monogr. Soc. Res. Child. Dev. 2013, 78, 49–69. [Google Scholar] [CrossRef]
- Glasser, M.F.; Sotiropoulos, S.N.; Wilson, J.A.; Coalson, T.S.; Fischl, B.; Andersson, J.L.; Xu, J.; Jbabdi, S.; Webster, M.; Polimeni, J.R.; et al. The minimal preprocessing pipelines for the Human Connectome Project. Neuroimage 2013, 80, 105–124. [Google Scholar] [CrossRef]
- Heister, D.; Brewer, J.B.; Magda, S.; Blennow, K.; McEvoy, L.K. Alzheimer’s Disease Neuroimaging Initiative Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology 2011, 77, 1619–1628. [Google Scholar] [CrossRef]
- Jenkinson, M.; Beckmann, C.F.; Behrens, T.E.; Woolrich, M.W.; Smith, S.M. FSL. Neuroimage 2012, 62, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.L.; Skare, S.; Ashburner, J. How to correct susceptibility distortions in spin-echo echo-planar images: Application to diffusion tensor imaging. Neuroimage 2003, 20, 870–888. [Google Scholar] [CrossRef]
- Griffanti, L.; Salimi-Khorshidi, G.; Beckmann, C.F.; Auerbach, E.J.; Douaud, G.; Sexton, C.E.; Zsoldos, E.; Ebmeier, K.P.; Filippini, N.; Mackay, C.E.; et al. ICA-based artefact removal and accelerated fMRI acquisition for improved resting state network imaging. Neuroimage 2014, 95, 232–247. [Google Scholar] [CrossRef]
- Salimi-Khorshidi, G.; Douaud, G.; Beckmann, C.F.; Glasser, M.F.; Griffanti, L.; Smith, S.M. Automatic denoising of functional MRI data: Combining independent component analysis and hierarchical fusion of classifiers. Neuroimage 2014, 90, 449–468. [Google Scholar] [CrossRef] [PubMed]
- Pruim, R.H.R.; Mennes, M.; Buitelaar, J.K.; Beckmann, C.F. Evaluation of ICA-AROMA and alternative strategies for motion artifact removal in resting state fMRI. Neuroimage 2015, 112, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dougherty, J.H.; Hubner, K.F.; Bai, B.; Cannon, R.L.; Hutson, R.K. Abnormal connectivity in the posterior cingulate and hippocampus in early Alzheimer’s disease and mild cognitive impairment. Alzheimers Dement. 2008, 4, 265–270. [Google Scholar] [CrossRef]
- Cox, R.W. AFNI: Software for analysis and visualization of functional magnetic resonance neuroimages. Comput. Biomed. Res. 1996, 29, 162–173. [Google Scholar] [CrossRef]
- D’Agostino, R.B., Jr. Tutorial in biostatistics: Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat. Med. 1998, 17, 2265–2281. [Google Scholar] [CrossRef]
- Brookhart, M.A.; Wyss, R.; Layton, J.B.; Stürmer, T. Propensity score methods for confounding control in nonexperimental research. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 604–611. [Google Scholar] [CrossRef]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. MatchIt: Nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 2011, 42, 1–28. [Google Scholar] [CrossRef]
- Gu, X.S.; Rosenbaum, P.R. Comparison of multivariate matching methods: Structures, distances, and algorithms. J. Comput. Graph. Stat. 1993, 2, 405–420. [Google Scholar]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Rugg, M.D.; Vilberg, K.L. Brain networks underlying episodic memory retrieval. Curr. Opin. Neurobiol. 2013, 23, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.; Mormino, E.; Johnson, K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef]
- Song, Z.; McDonough, I.M.; Liu, P.; Lu, H.; Park, D.C. Cortical amyloid burden and age moderate hippocampal activity in cognitively-normal adults. Neuroimage Clin. 2016, 12, 78–84. [Google Scholar] [CrossRef]
- Jones, D.T.; Knopman, D.S.; Gunter, J.L.; Graff-Radford, J.; Vemuri, P.; Boeve, B.F.; Petersen, R.C.; Weiner, M.W.; Jack, C.R., Jr. Cascading network failure across the Alzheimer’s disease spectrum. Brain 2016, 139, 547–562. [Google Scholar] [CrossRef]
- Grady, C.L.; Garrett, D.D. Understanding variability in the BOLD signal and why it matters for aging. Brain Imaging Behav. 2014, 8, 274–283. [Google Scholar] [CrossRef]
- Zöller, D.; Schaer, M.; Scariati, E.; Padula, M.C.; Eliez, S.; Van De Ville, D. Disentangling resting-state BOLD variability and PCC functional connectivity in 22q11.2 deletion syndrome. Neuroimage 2017, 149, 85–97. [Google Scholar] [CrossRef]
- Ramírez-Toraño, F.; Abbas, K.; Bruña, R.; Marcos de Pedro, S.; Gómez-Ruiz, N.; Barabash, A.; Pereda, E.; Marcos, A.; López-Higes, R.; Maestu, F.; et al. A Structural Connectivity Disruption One Decade before the Typical Age for Dementia: A Study in Healthy Subjects with Family History of Alzheimer’s Disease. Cereb. Cortex Commun. 2021, 2, tgab051. [Google Scholar] [CrossRef]
- McDonough, I.M.; Letang, S.K.; Stinson, E.A. Dementia risk elevates brain activity during memory retrieval: A functional MRI analysis of middle aged and older adults. J. Alzheimer Dis. 2019, 70, 1005–1023. [Google Scholar] [CrossRef] [PubMed]
- D’Esposito, M.; Deouell, L.Y.; Gazzaley, A. Alterations in the BOLD fMRI signal with ageing and disease: A challenge for neuroimaging. Nat. Rev. Neurosci. 2003, 4, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.L.; Kwong, K.K.; Weisskoff, R.M.; Rosen, B.R. Calibrated functional MRI: Mapping the dynamics of oxidative metabolism. Proc. Natl. Acad. Sci. USA 1998, 95, 1834–1839. [Google Scholar] [CrossRef]
- Gonneaud, J.; Baria, A.T.; Pichet Binette, A.; Gordon, B.A.; Chhatwal, J.P.; Cruchaga, C.; Jucker, M.; Levin, J.; Salloway, S.; Farlow, M.; et al. Accelerated functional brain aging in pre-clinical familial Alzheimer’s disease. Nat. Commun. 2021, 12, 5346. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parent without AD | Parent with AD | |
|---|---|---|
| N | 14 | 14 |
| Age (years) | 28.93 (4.34) | 29.86 (4.62) |
| Age Range | 22–35 | 22–35 |
| Known APOE E4 Positive (N) | 2 | 4 |
| Sex (M/F) | 6/8 | 7/7 |
| Race (White) | 79% | 79% |
| Ethnicity (Hispanic) | 7% | 7% |
| Education (years) | 13.79 (1.67) | 13.86 (1.96) |
| Education Range | 11–17 | 11–17 |
| Employment Status | ||
| Not Working | 14% | 36% |
| Part Time | 43% | 14% |
| Full Time | 43% | 50% |
| Parents with Depression | 71% | 71% |
| Parents with Bipolar Disorder | 36% | 29% |
| Mini_Mental State Exam Score | 29.29 (0.91) | 29.00 (0.96) |
| Mini_Mental State Exam Range | 27–30 | 27–30 |
| Reading Ability Score | 116.04 (7.31) | 116.50 (11.62) |
| Reading Ability Range | 103.51–141.32 | 100.63–141.32 |
| Verbal Memory Score | 35.21 (2.91) | 34.92 (3.34) |
| Verbal Memory Range | 30–39 | 30–39 |
| Visual Memory Score | 108.60 (9.70) | 107.07 (10.99) |
| Visual Memory Range | 88.97–121.82 | 87.10–125.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonough, I.M.; Mayhugh, C.; Moore, M.K.; Brasfield, M.B.; Letang, S.K.; Madan, C.R.; Allen, R.S. Young Adults with a Parent with Dementia Show Early Abnormalities in Brain Activity and Brain Volume in the Hippocampus: A Matched Case-Control Study. Brain Sci. 2022, 12, 496. https://doi.org/10.3390/brainsci12040496
McDonough IM, Mayhugh C, Moore MK, Brasfield MB, Letang SK, Madan CR, Allen RS. Young Adults with a Parent with Dementia Show Early Abnormalities in Brain Activity and Brain Volume in the Hippocampus: A Matched Case-Control Study. Brain Sciences. 2022; 12(4):496. https://doi.org/10.3390/brainsci12040496
Chicago/Turabian StyleMcDonough, Ian M., Christopher Mayhugh, Mary Katherine Moore, Mikenzi B. Brasfield, Sarah K. Letang, Christopher R. Madan, and Rebecca S. Allen. 2022. "Young Adults with a Parent with Dementia Show Early Abnormalities in Brain Activity and Brain Volume in the Hippocampus: A Matched Case-Control Study" Brain Sciences 12, no. 4: 496. https://doi.org/10.3390/brainsci12040496
APA StyleMcDonough, I. M., Mayhugh, C., Moore, M. K., Brasfield, M. B., Letang, S. K., Madan, C. R., & Allen, R. S. (2022). Young Adults with a Parent with Dementia Show Early Abnormalities in Brain Activity and Brain Volume in the Hippocampus: A Matched Case-Control Study. Brain Sciences, 12(4), 496. https://doi.org/10.3390/brainsci12040496