CNS Superficial Siderosis Mimicking a Motor Neuron Disease

 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
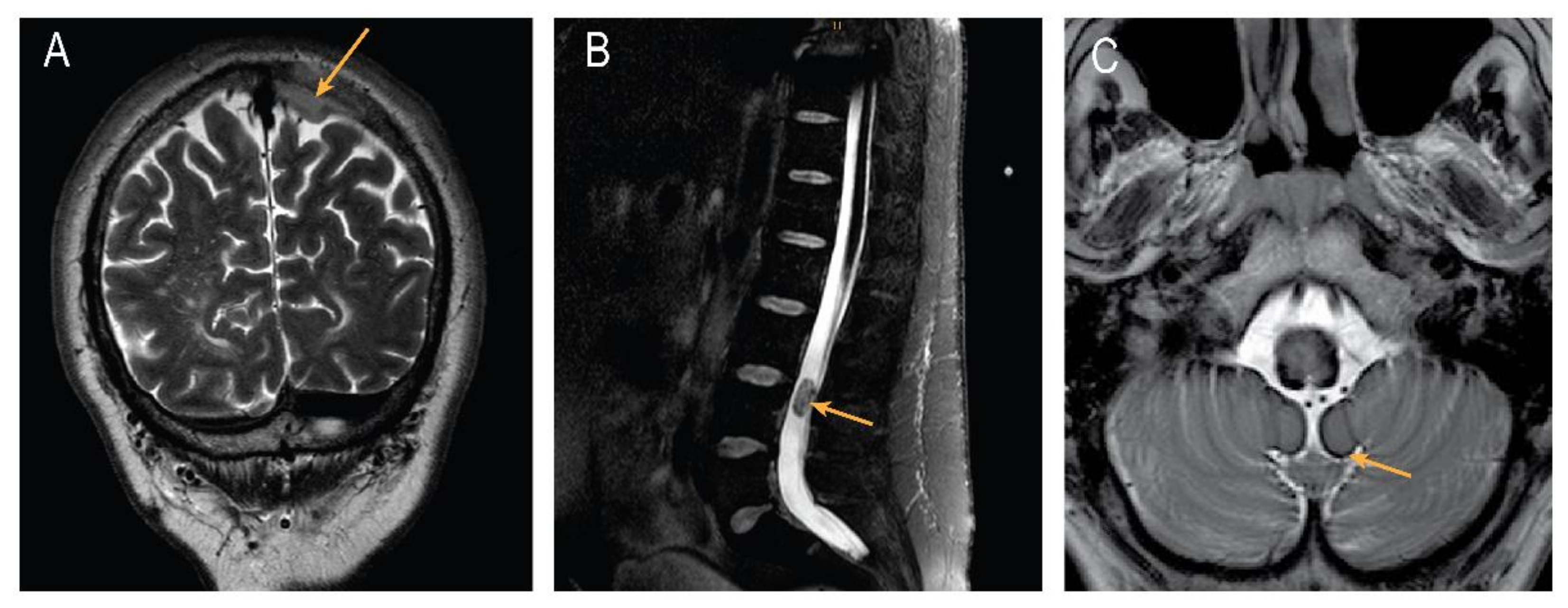
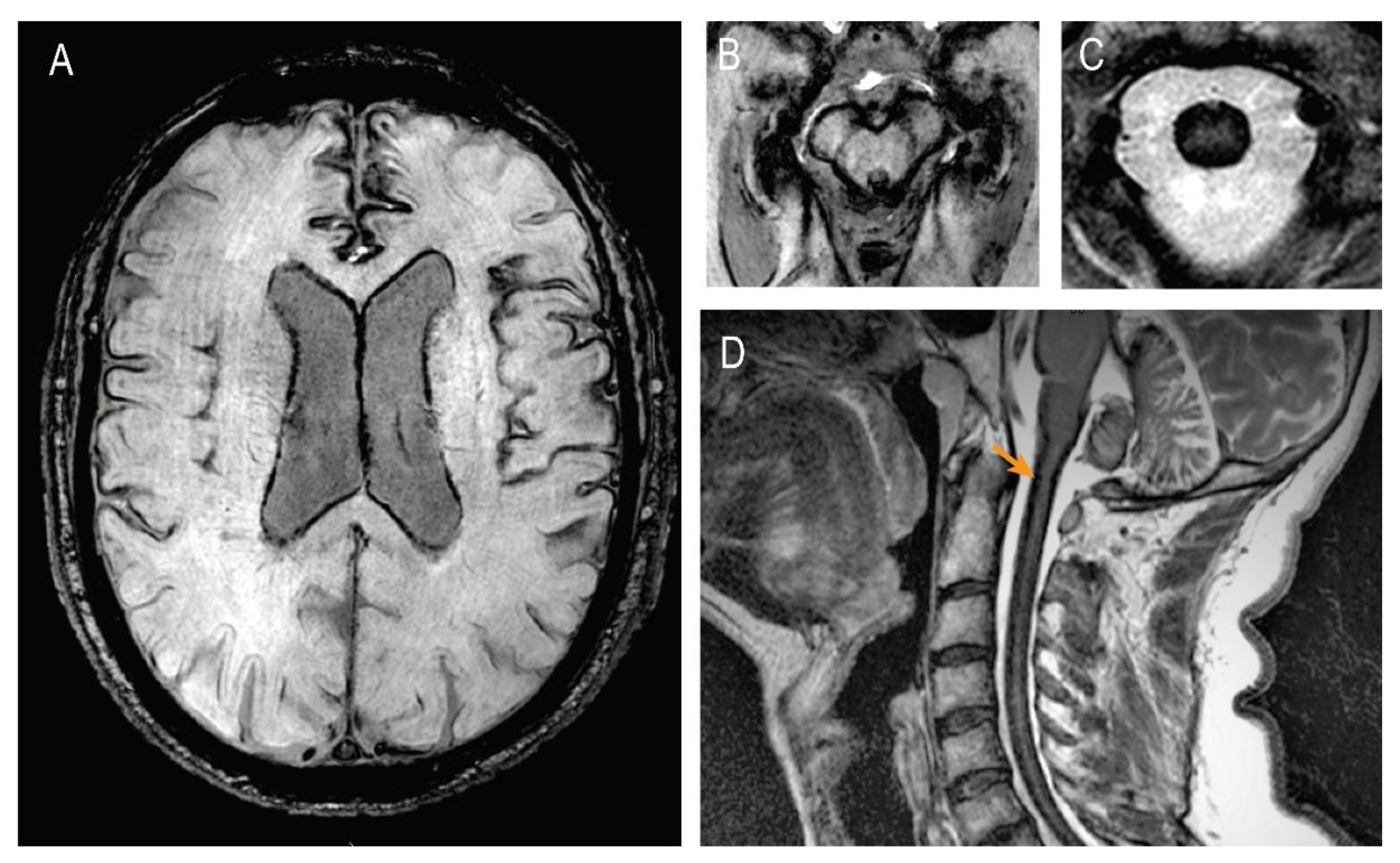
2. Case Presentation

3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fearnley, J.M.; Stevens, J.M.; Rudge, P. Superficial Siderosis of the Central Nervous System. Brain 1995, 118, 1051–1066. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Cohen-Gadol, A.A.; Wright, R.A.; Miller, G.M.; Piepgras, D.G.; Ahlskog, J.E. Superficial Siderosis. Neurology 2006, 66, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Vemuri, P.; Rabinstein, A.A.; Aakre, J.; Flemming, K.D.; Brown, R.D.; Kumar, N.; Kantarci, K.; Kremers, W.; Mielke, M.M.; et al. Prevalence and Natural History of Superficial Siderosis: A Population-Based Study. Stroke 2017, 48, 3210–3214. [Google Scholar] [CrossRef] [PubMed]
- Leussink, V.I.; Flachenecker, P.; Brechtelsbauer, D.; Bendszus, M.; Sliwka, U.; Gold, R.; Becker, G. Superficial Siderosis of the Central Nervous System: Pathogenetic Heterogeneity and Therapeutic Approaches. Acta Neurol Scand 2003, 107, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N. Beyond Superficial Siderosis: Introducing “Duropathies. ” Neurology 2012, 78, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Vicente, E.; Pradas, J.; Marín-lahoz, J.; De Luna, N.; Clarimón, J.; Turon-Sans, J.; Gelpí, E.; Díaz-Manera, J.; Illa, I.; Rojas-Garcia, R. Early Diagnosis of Amyotrophic Lateral Sclerosis Mimic Syndromes: Pros and Cons of Current Clinical Diagnostic Criteria. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Turner, B. Superficial Siderosis Associated with Anterior Horn Cell Dysfunction. J. Neurol. Neurosurg. Psychiatry 2002, 72, 275. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Driver-Dunckley, E.D.; Hoxworth, J.M.; Patel, N.P.; Bosch, E.P.; Goodman, B.P. Superficial Siderosis Mimicking Amyotrophic Lateral Sclerosis. J. Clin. Neuromuscul. Dis. 2010, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Payer, M.; Sottas, C.; Bonvin, C. Superficial Siderosis of the Central Nervous System: Secondary Progression despite Successful Surgical Treatment, Mimicking Amyotrophic Lateral Sclerosis. Case Report and Review. Acta Neurochir. 2010, 152, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Fogelson, J.L.; Morris, J.M.; Pichelmann, M.A. Superficial Siderosis Should Be Included in the Differential Diagnosis of Motor Neuron Disease. Neurologist 2012, 18, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, K.; Honjo, N.; Takata, T.; Touge, T.; Masaki, T. Flail Arm Syndrome Mimic Caused by Hemosiderin Deposition in the Anterior Horn. Acta Neurol. Belg. 2020, 120, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.A.; Li, X.; Schwartz, K.; Huang, H.; Mealy, M.A.; Levy, M. Two-Year Observational Study of Deferiprone in Superficial Siderosis. CNS Neurosci. Ther. 2018, 24, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Michael, S.C.; Li, D.; Chen, Z.; Cusack, M.J.; Gibson, W.M.; Petrocine, S.V.; Qian, J. The Pathology of Superficial Siderosis of the Central Nervous System. Acta Neuropathol. 2008, 116, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T. Microglia Take Centre Stage in Neurodegenerative Disease. Nat. Rev. Immunol. 2019, 19, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Dickson, A.C. Tin-Protoporphyrin Prevents Experimental Superficial Siderosis in Rabbits. J. Neuropathol. Exp. Neurol. 2002, 61, 689–701. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Gomez, S.; Binder, J.; Schievelkamp, A.-H.; Heneka, M.T. CNS Superficial Siderosis Mimicking a Motor Neuron Disease. Brain Sci. 2022, 12, 1558. https://doi.org/10.3390/brainsci12111558
Castro-Gomez S, Binder J, Schievelkamp A-H, Heneka MT. CNS Superficial Siderosis Mimicking a Motor Neuron Disease. Brain Sciences. 2022; 12(11):1558. https://doi.org/10.3390/brainsci12111558
Chicago/Turabian StyleCastro-Gomez, Sergio, Julius Binder, Arndt-Hendrik Schievelkamp, and Michael Thomas Heneka. 2022. "CNS Superficial Siderosis Mimicking a Motor Neuron Disease" Brain Sciences 12, no. 11: 1558. https://doi.org/10.3390/brainsci12111558
APA StyleCastro-Gomez, S., Binder, J., Schievelkamp, A.-H., & Heneka, M. T. (2022). CNS Superficial Siderosis Mimicking a Motor Neuron Disease. Brain Sciences, 12(11), 1558. https://doi.org/10.3390/brainsci12111558