
Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
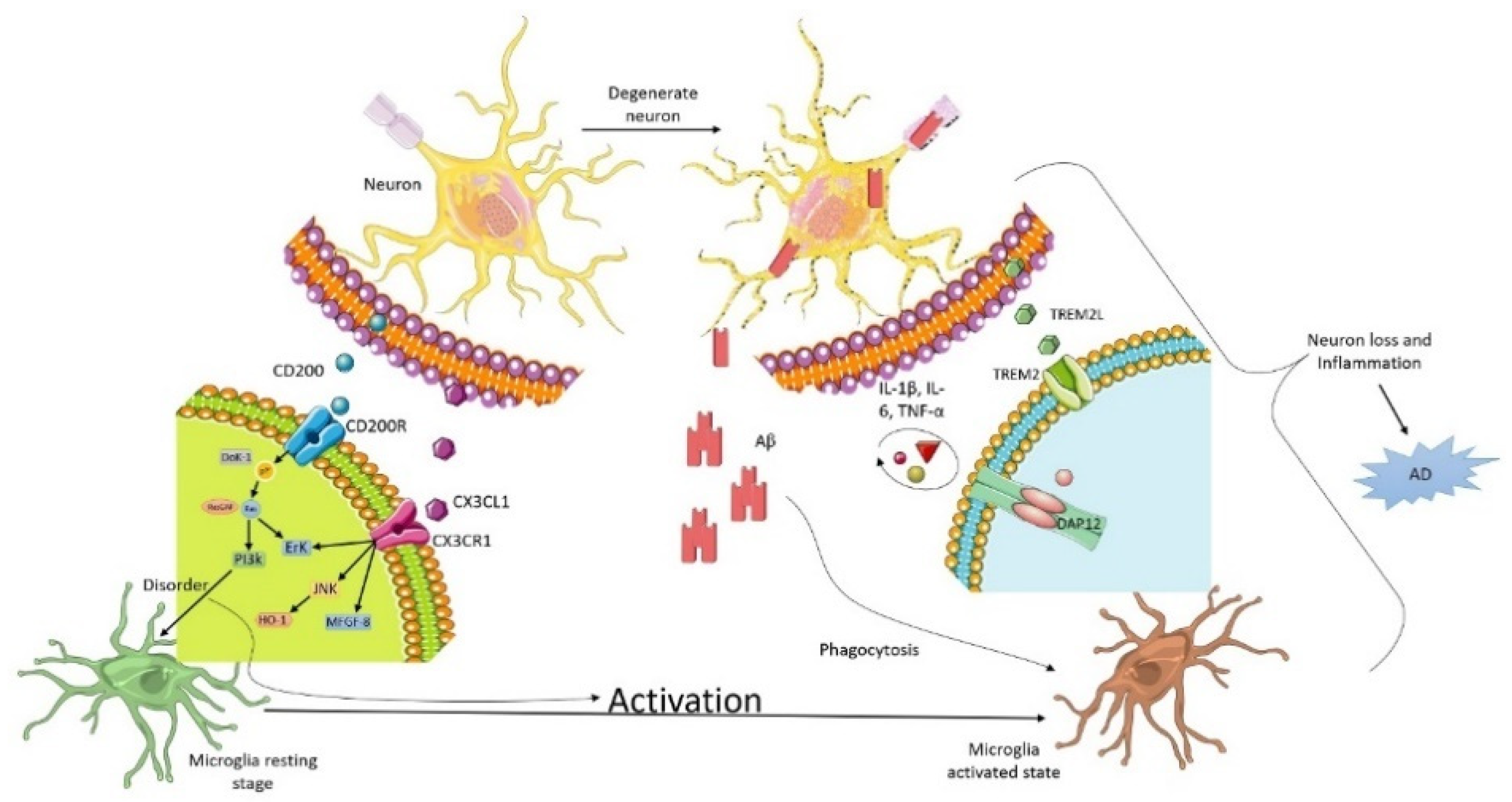
2. Microglia and Neuron Interaction in Alzheimer
3. Triggering Receptors Expressed on Myeloid Cell-2 (TREM2) Receptor
3.1. CD200
3.2. CX3XL1
3.3. Extra-Internal Neuronal Signaling Pathways
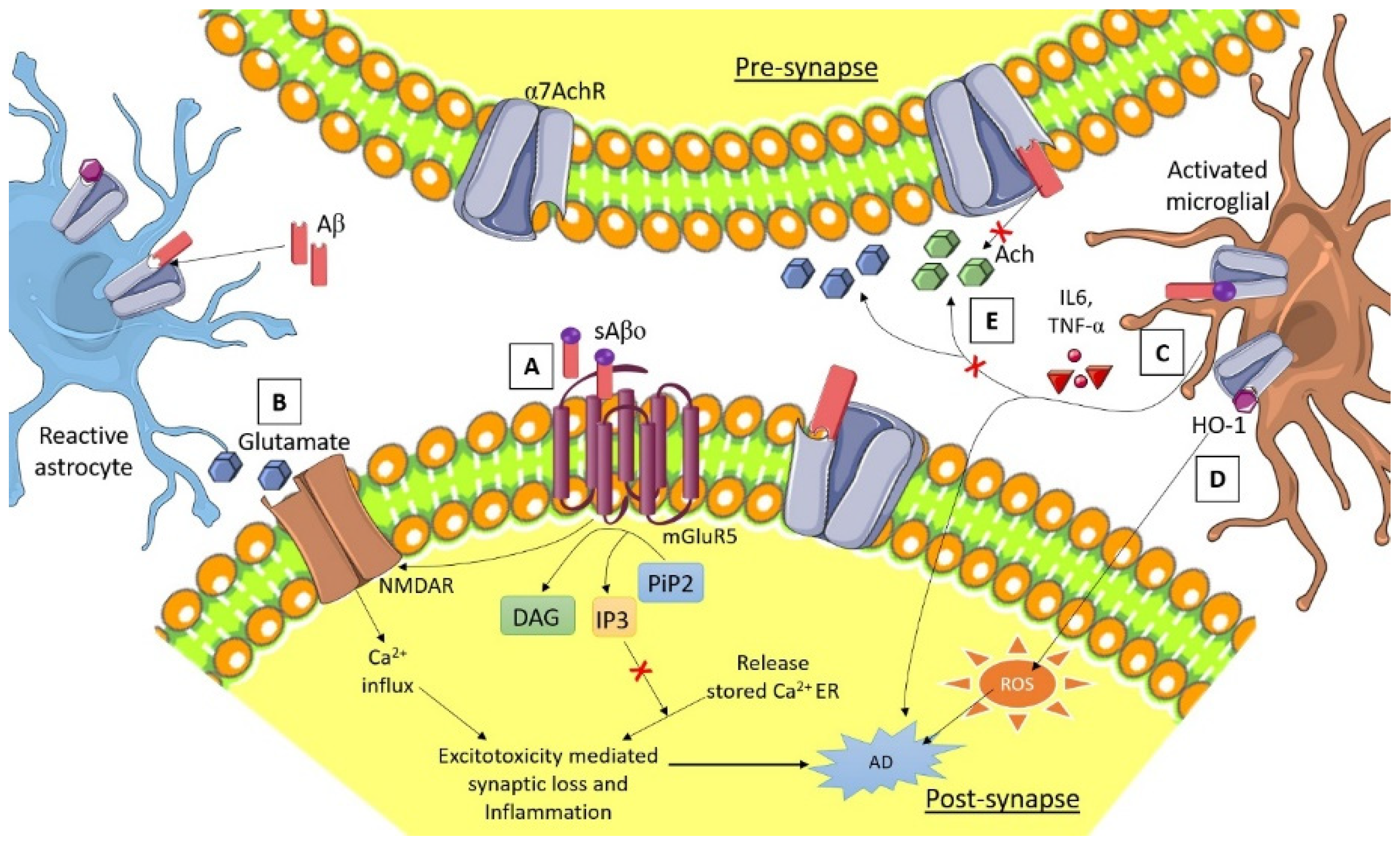
3.4. Astrocytes and Neuron Interaction in AD
3.5. Contradiction of Metabolic Lactate Transition between Astrocytes and Neurons
3.6. Calcium Homeostasis
3.7. α7 Nicotinic Acetylcholine Receptor (α7nAChRs) Synaptic Function in AD Pathology
3.8. Microglia and Astrocytes
3.9. Oligodendrocytes
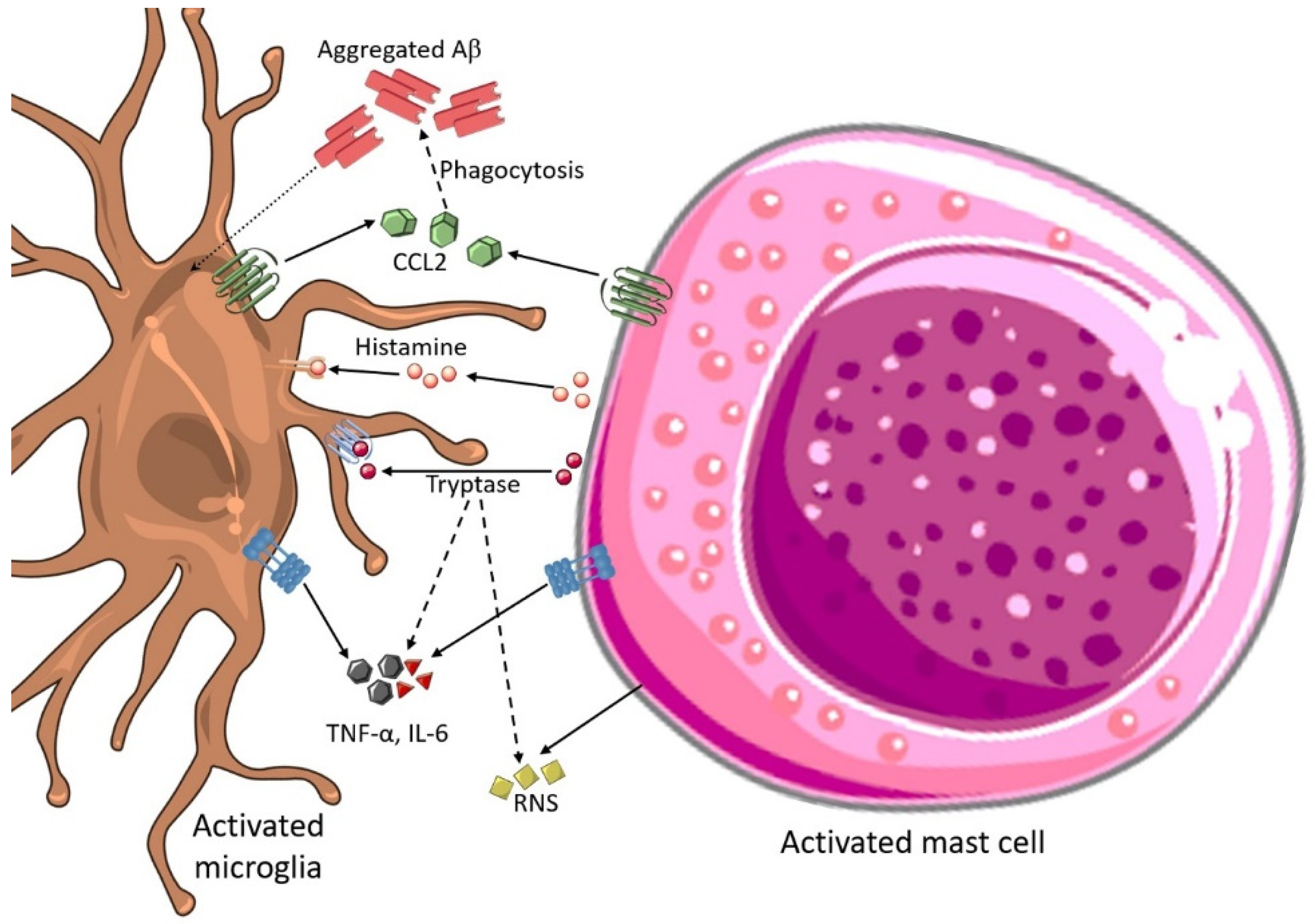
3.10. Mast Cell and Microglial Interactions in AD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Salmina, A.B. Neuron-glia interactions as therapeutic targets in neurodegeneration. J. Alzheimers Dis. 2009, 16, 485–502. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, M.H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron-Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]
- Abbott, A. Dementia: A problem for our age. Nature 2011, 475, S2–S4. [Google Scholar] [CrossRef]
- Sosa-Ortiz, A.L.; Acosta-Castillo, I.; Prince, M.J. Epidemiology of dementias and Alzheimer’s disease. Arch. Med. Res. 2012, 43, 600–608. [Google Scholar] [CrossRef]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Min, R.; Santello, M.; Nevian, T. The computational power of astrocyte mediated synaptic plasticity. Front. Comput. Neurosci. 2012, 6, 93. [Google Scholar] [CrossRef]
- Garcia-Segura, L.M.; Balthazart, J. Steroids and neuroprotection: New advances. Front. Neuroendocrinol. 2009, 30, v–ix. [Google Scholar] [CrossRef]
- Watkins, L.R.; Milligan, E.D.; Maier, S.F. Glial activation: A driving force for pathological pain. Trends Neurosci. 2001, 24, 450–455. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Hong, H.; Kim, B.S.; Im, H.I. Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int. Neurourol. J. 2016, 20, S2–S7. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Davis, A.A.; Leyns, C.E.G.; Holtzman, D.M. Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 545–568. [Google Scholar] [CrossRef]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Puranik, N.; Yadav, D.; Yadav, S.K.; Chavda, V.K.; Jin, J.O. Proteomics and Neurodegenerative Disorders: Advancements in the Diagnostic Analysis. Curr. Protein Pept. Sci. 2020, 21, 1174–1183. [Google Scholar] [CrossRef]
- Coppede, F.; Mancuso, M.; Siciliano, G.; Migliore, L.; Murri, L. Genes and the environment in neurodegeneration. Biosci. Rep. 2006, 26, 341–367. [Google Scholar] [CrossRef]
- Pandey, M.; Nabi, J.; Tabassum, N.; Pottoo, F.H.; Khatik, R.; Ahmad, N. Molecular chaperones in neurodegeneration: Mechanisms of regulation in cellular proteostasis. In Quality Control of Cellular Protein in Neurodegenerative Disorders; Uddin, S., Ashraf, G.M., Eds.; IGI Global: Hershey, PA, USA, 2020; pp. 354–379. [Google Scholar]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef]
- Singh, K.; Yadav, D.; Chauhan, P.S.; Mishra, M.; Jin, J.O. Novel Therapeutics for the Treatment of Alzheimer’s and Parkinson’s Disease. Curr. Pharm. Des. 2020, 26, 755–763. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Benarroch, E.E. Neuron-astrocyte interactions: Partnership for normal function and disease in the central nervous system. Mayo Clin. Proc. 2005, 80, 1326–1338. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk between astrocytes and microglia: An overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Fellin, T.; Carmignoto, G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J. Physiol. 2004, 559, 3–15. [Google Scholar] [CrossRef]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia crosstalk in neuroinflammatory diseases. Front. Cell. Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef]
- Kery, R.; Chen, A.P.; Kirschen, G.W. Genetic targeting of astrocytes to combat neurodegenerative disease. Neural Regen. Res. 2020, 15, 199. [Google Scholar]
- Bernier, L.-P.; York, E.M.; MacVicar, B.A. Immunometabolism in the brain: How metabolism shapes microglial function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef]
- Gupta, N.; Shyamasundar, S.; Patnala, R.; Karthikeyan, A.; Arumugam, T.V.; Ling, E.-A.; Dheen, S.T. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin. Ther. Targets 2018, 22, 765–781. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T. Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer’s disease model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 2018, 98, 1170–1183.e1178. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the central nervous system. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Zorec, R.; Županc, T.A.; Verkhratsky, A. Astrogliopathology in the infectious insults of the brain. Neurosci. Lett. 2019, 689, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, Q.; Zhao, C.; Da, Y.; Zhang, H.-L.; Chen, Z. Differentiation of Glial Cells From hiPSCs: Potential Applications in Neurological Diseases and Cell Replacement Therapy. Front. Cell. Neurosci. 2018, 12, 239. [Google Scholar] [CrossRef]
- Kettenmann, H.; Verkhratsky, A. Neuroglia: The 150 years after. Trends Neurosci. 2008, 31, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 2018, 48, 380–395.e386. [Google Scholar] [CrossRef]
- Amor, S.; Woodroofe, M.N. Innate and adaptive immune responses in neurodegeneration and repair. Immunology 2014, 141, 287–291. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Sheridan, G.K.; Murphy, K.J. Neuron–glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef]
- Barclay, A.N.; Wright, G.J.; Brooke, G.; Brown, M.H. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002, 23, 285–290. [Google Scholar] [CrossRef]
- Costantini, E.; D’Angelo, C.; Reale, M. The role of immunosenescence in neurodegenerative diseases. Mediat. Inflamm. 2018, 2018, 6039171. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.-F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Downer, E.J.; Crotty, S.; Nolan, Y.M.; Mills, K.H.; Lynch, M.A. CD200 ligand–receptor interaction modulates microglial activation in vivo and in vitro: A role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Milligan, E.; Sloane, E.; Watkins, L. Glia in pathological pain: A role for fractalkine. J. Neuroimmunol. 2008, 198, 113–120. [Google Scholar] [CrossRef]
- Lauro, C.; Catalano, M.; Trettel, F.; Limatola, C. Fractalkine in the nervous system: Neuroprotective or neurotoxic molecule. Ann. N. Y. Acad. Sci. 2015, 1351, 141–148. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, Q.; Song, D.; Quan, Z.; Qing, H. Optogenetic manipulation of astrocytes from synapses to neuronal networks: A potential therapeutic strategy for neurodegenerative diseases. Glia 2020, 68, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.; Vuillaume, R.; Binczak, S.; Jacquir, S. Spatiotemporal model of tripartite synapse with perinodal astrocytic process. J. Comput. Neurosci. 2020, 48, 1–20. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-protein-coupled receptors in astrocyte–neuron communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; Morley, J.W.; Buskila, Y. Neuromodulation of glial function during neurodegeneration. Front. Cell. Neurosci. 2020, 14, 278. [Google Scholar] [CrossRef]
- Bellot-Saez, A.; Stevenson, R.; Kekesi, O.; Samokhina, E.; Ben-Abu, Y.; Morley, J.W.; Buskila, Y. Neuromodulation of astrocytic K+ clearance. Int. J. Mol. Sci. 2021, 22, 2520. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, C.; Weiss, G.L.; Fu, X.; Fisher, M.O.; Begley, J.C.; Stevens, C.R.; Harrison, L.M.; Tasker, J.G. Acute stress desensitizes hypothalamic CRH neurons to norepinephrine and physiological stress. bioRxiv 2020. [Google Scholar] [CrossRef]
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef]
- Quandt, F.; MacVicar, B. Calcium activated potassium channels in cultured astrocytes. Neuroscience 1986, 19, 29–41. [Google Scholar] [CrossRef]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef]
- Alexander, S.P.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J. The concise guide to pharmacology 2021/22: Ion channels. Br. J. Pharmacol. 2021, 178, S157–S245. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Gordon, G.R.; Mulligan, S.J.; MacVicar, B.A. Astrocyte control of the cerebrovasculature. Glia 2007, 55, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef]
- Raskin, J.; Cummings, J.; Hardy, J.; Schuh, K.; Dean, R.A. Neurobiology of Alzheimer’s disease: Integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr. Alzheimer Res. 2015, 12, 712–722. [Google Scholar] [CrossRef]
- Shi, Z.M.; Han, Y.W.; Han, X.H.; Zhang, K.; Chang, Y.N.; Hu, Z.M.; Qi, H.X.; Ting, C.; Zhen, Z.; Hong, W. Upstream regulators and downstream effectors of NF-kappaB in Alzheimer’s disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef]
- Hou, L.; Liu, Y.; Wang, X.; Ma, H.; He, J.; Zhang, Y.; Yu, C.; Guan, W.; Ma, Y. The effects of amyloid-beta42 oligomer on the proliferation and activation of astrocytes in vitro. In Vitro Cell. Dev. Biol. Anim. 2011, 47, 573–580. [Google Scholar] [CrossRef]
- Albrecht, J.; Szeliga, M.; Zielińska, M. Brain Glutamine: Roles in Norm and Pathology. In Glutamine; CRC Press: Boca Raton, FL, USA, 2017; pp. 95–110. [Google Scholar]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Dashkoff, J.; Roe, A.D.; Takeda, S.; Koffie, R.M.; Hashimoto, T.; Scheel, M.; Spires-Jones, T.; Arbel-Ornath, M.; Betensky, R.; et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 2013, 5, 212ra161. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Deng, A.; Lleo, A.; Berezovska, O.; Von Arnim, C.A.; Martin-Rehrmann, M.; Manelli, A.; LaDu, M.J.; Hyman, B.T.; Rebeck, G.W. Apolipoprotein E modulates gamma-secretase cleavage of the amyloid precursor protein. J. Neurochem. 2004, 90, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, J.S.; Gomes, C.M. S100 proteins in Alzheimer’s disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S. Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 273–279. [Google Scholar] [CrossRef]
- Hertz, L.; Zielke, H.R. Astrocytic control of glutamatergic activity: Astrocytes as stars of the show. Trends Neurosci. 2004, 27, 735–743. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef]
- Amara, S.G.; Fontana, A.C. Excitatory amino acid transporters: Keeping up with glutamate. Neurochem. Int. 2002, 41, 313–318. [Google Scholar] [CrossRef]
- Sonnewald, U.; Qu, H.; Aschner, M. Pharmacology and toxicology of astrocyte-neuron glutamate transport and cycling. J. Pharmacol. Exp. Ther. 2002, 301, 1–6. [Google Scholar] [CrossRef]
- Leal, M.C.; Dorfman, V.B.; Gamba, A.F.; Frangione, B.; Wisniewski, T.; Castano, E.M.; Sigurdsson, E.M.; Morelli, L. Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J. Neuropathol. Exp. Neurol. 2006, 65, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Zipfel, P.; Rochais, C.; Baranger, K.; Rivera, S.; Dallemagne, P. Matrix metalloproteinases as new targets in Alzheimer’s disease: Opportunities and challenges. J. Med. Chem. 2020, 63, 10705–10725. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef] [PubMed]
- Chih, C.-P.; Lipton, P.; Roberts, E.L. Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci. 2001, 24, 573–578. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Pellerin, L. Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol. Sci. 1999, 14, 177–182. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.; Singh, S.K. The astrocyte–neuron interface: An overview on molecular and cellular dynamics controlling formation and maintenance of the tripartite synapse. In Methods in Molecular Biology; Benedetto, B., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1938, pp. 3–18. [Google Scholar]
- Garwood, C.; Faizullabhoy, A.; Wharton, S.B.; Ince, P.G.; Heath, P.R.; Shaw, P.J.; Baxter, L.; Gelsthorpe, C.; Forster, G.; Matthews, F.E.; et al. Calcium dysregulation in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 2013, 39, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca(2+) homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef]
- Rudy, C.C.; Hunsberger, H.C.; Weitzner, D.S.; Reed, M.N. The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer’s disease. Aging Dis. 2015, 6, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Majdi, A.; Mahmoudi, J.; Golzari, S.E.J.; Talebi, M. Astrocytic and microglial nicotinic acetylcholine receptors: An overlooked issue in Alzheimer’s disease. J. Neural Transm. 2016, 123, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant calcium signals in reactive astrocytes: A key process in neurological disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Zhang, W.J.; Su, D.F.; Zhang, G.Q.; Miao, C.Y. Cellular responses and functions of alpha7 nicotinic acetylcholine receptor activation in the brain: A narrative review. Ann. Transl. Med. 2021, 9, 509. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M.; Golzari, S.E.J.; Sabermarouf, B.; Mahmoudi, J. Beta-amyloid exhibits antagonistic effects on alpha 7 nicotinic acetylcholine receptors in orchestrated manner. J. Med. Hypotheses Ideas 2014, 8, 49–52. [Google Scholar] [CrossRef]
- Echeverria, V.; Yarkov, A.; Aliev, G. Positive modulators of the alpha7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 142–157. [Google Scholar] [CrossRef]
- Kalashnyk, O.; Lykhmus, O.; Oliinyk, O.; Komisarenko, S.; Skok, M. alpha7 Nicotinic acetylcholine receptor-specific antibody stimulates interleukin-6 production in human astrocytes through p38-dependent pathway. Int. Immunopharmacol. 2014, 23, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Talebi, M.; Mahmoudi, J.; Babri, S.; Shanehbandi, D. Selective activation of alpha7 nicotinic acetylcholine receptor by PHA-543613 improves Abeta25-35-mediated cognitive deficits in mice. Neuroscience 2015, 298, 81–93. [Google Scholar] [CrossRef]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Foucault-Fruchard, L.; Antier, D. Therapeutic potential of alpha7 nicotinic receptor agonists to regulate neuroinflammation in neurodegenerative diseases. Neural Regen. Res. 2017, 12, 1418–1421. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.Y.; DeLeo, J.A. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur. J. Neurosci. 2004, 20, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Weaver-Mikaere, L.; Gunn, A.J.; Mitchell, M.D.; Bennet, L.; Fraser, M. LPS and TNF alpha modulate AMPA/NMDA receptor subunit expression and induce PGE2 and glutamate release in preterm fetal ovine mixed glial cultures. J. Neuroinflammation 2013, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, M.K.; Paintlia, A.S.; Khan, M.; Singh, I.; Singh, A.K. Modulation of peroxisome proliferator-activated receptor-α activity by N-acetyl cysteine attenuates inhibition of oligodendrocyte development in lipopolysaccharide stimulated mixed glial cultures. J. Neurochem. 2008, 105, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef]
- Márquez-Ropero, M.; Benito, E.; Plaza-Zabala, A.; Sierra, A. Microglial Corpse Clearance: Lessons From Macrophages. Front. Immunol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Chavda, V.; Lu, P.D.B.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G. Molecular Mechanisms of Oxidative stress in Stroke and Cancer. Brain Disord. 2021, 5, 100029. [Google Scholar] [CrossRef]
- Khurana, M.; Rahman, S.O.; Najmi, A.K.; Pottoo, F.H.; Akhtar, M.S. Application of Contemporary Neuroproteomic Techniques in Unravelling Neurological Disorders. Curr Protein Pept Sci. 2020, 21, 1146–1163. [Google Scholar] [CrossRef]
- Varas, R.; Ortiz, F.C. Neuroinflammation in demyelinating diseases: Oxidative stress as a modulator of glial cross-talk. Curr. Pharm. Des. 2019, 25, 4755–4762. [Google Scholar] [CrossRef]
- Nicol, B.; Salou, M.; Laplaud, D.A.; Wekerle, H. The autoimmune concept of multiple sclerosis. Presse Med. 2015, 44, e103–e112. [Google Scholar] [CrossRef]
- Leviton, A.; Gressens, P. Neuronal damage accompanies perinatal white-matter damage. Trends Neurosci. 2007, 30, 473–478. [Google Scholar] [CrossRef]
- Strunk, T.; Inder, T.; Wang, X.; Burgner, D.; Mallard, C.; Levy, O. Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect. Dis. 2014, 14, 751–762. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Aboul-Enein, F.; Rauschka, H.; Kornek, B.; Stadelmann, C.; Stefferl, A.; Brück, W.; Lucchinetti, C.; Schmidbauer, M.; Jellinger, K.; Lassmann, H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol. 2003, 62, 25–33. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef]
- Franklin, R.J.; Goldman, S.A. Glia Disease and Repair-Remyelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020594. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Zaheer, S.; Thangavel, R.; Ahmed, M.E.; Raikwar, S.; Govindarajan, R.; Iyer, S.; Zaheer, A. Cross-talk between glia, neurons and mast cells in neuroinflammation associated with Parkinson’s disease. J. Neuroimmune Pharmacol. 2018, 13, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Kulka, M. Decoding mast cell-microglia communication in neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.; Rapanelli, M.; Abbasi, E.; Ohtsu, H.; Pittenger, C. Histamine regulation of microglia: Gene-environment interaction in the regulation of central nervous system inflammation. Brain Behav. Immun. 2016, 57, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, W.; Zeng, X.; Hu, G.; Zhang, H.; He, S.; Zhang, S. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol. Neurobiol. 2014, 49, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Santos, T.; Goncalves, J.; Baltazar, G.; Ferreira, L.; Agasse, F.; Bernardino, L. Histamine modulates microglia function. J. Neuroinflammation 2012, 9, 90. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Dubova, I.; Giler, G.; et al. Mast cell proteases activate astrocytes and glia-neurons and release interleukin-33 by activating p38 and ERK1/2 MAPKs and NF-κB. Mol. Neurobiol. 2019, 56, 1681–1693. [Google Scholar] [CrossRef]
- Kiyota, T.; Yamamoto, M.; Xiong, H.; Lambert, M.P.; Klein, W.L.; Gendelman, H.E.; Ransohoff, R.M.; Ikezu, T. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS ONE 2009, 4, e6197. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Nair, A.; Gupta, M. Mast cells in neurodegenerative disease. Front. Cell. Neurosci. 2019, 13, 171. [Google Scholar] [CrossRef]
- Harcha, P.A.; Vargas, A.; Yi, C.; Koulakoff, A.A.; Giaume, C.; Saez, J.C. Hemichannels are required for amyloid β-peptide-induced degranulation and are activated in brain mast cells of APPswe/PS1dE9 mice. J. Neurosci. 2015, 35, 9526–9538. [Google Scholar] [CrossRef]
- Aguirre, A.; Maturana, C.J.; Harcha, P.A.; Sáez, J.C. Possible involvement of TLRs and hemichannels in stress-induced CNS dysfunction via mastocytes, and glia activation. Mediat. Inflamm. 2013, 2013, 893521. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavda, V.; Singh, K.; Patel, V.; Mishra, M.; Mishra, A.K. Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease. Brain Sci. 2022, 12, 75. https://doi.org/10.3390/brainsci12010075
Chavda V, Singh K, Patel V, Mishra M, Mishra AK. Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease. Brain Sciences. 2022; 12(1):75. https://doi.org/10.3390/brainsci12010075
Chicago/Turabian StyleChavda, Vishal, Kavita Singh, Vimal Patel, Meerambika Mishra, and Awdhesh Kumar Mishra. 2022. "Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease" Brain Sciences 12, no. 1: 75. https://doi.org/10.3390/brainsci12010075
APA StyleChavda, V., Singh, K., Patel, V., Mishra, M., & Mishra, A. K. (2022). Neuronal Glial Crosstalk: Specific and Shared Mechanisms in Alzheimer’s Disease. Brain Sciences, 12(1), 75. https://doi.org/10.3390/brainsci12010075