
SCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
Case Series
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliva, M.; Berkovic, S.F.; Petrou, S. Sodium channels and the neurobiology of epilepsy. Epilepsia 2012, 53, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Møller, R.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Linley, J.; Baker, M.D.; Minett, M.S.; Cregg, R.; Werdehausen, R.; Rugiero, F.; Wood, J.N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135 Pt 9, 2585–2612. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yasumoto, S.; Kurahashi, H.; Nakagawa, E.; Fukasawa, T.; Uchiya, S.; Hirose, S. Clinical spectrum of SCN2A mutations. Brain Dev. 2012, 34, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Du, J.; Steckler, F.; Ghanty, I.I.; Johannesen, K.M.; Fenger, C.D.; Schorge, S.; Baez-Nieto, D.; Wang, H.; Allen, A.; et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 2020, 61, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 15, 958–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdò, S.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindy, A.S.; Johannesen, K.M.; Hedrich, U.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, E.; Raviglione, F.; Franceschetti, S.; D’Arrigo, S.; Milani, D.; Selicorni, A.; Riva, D.; Zuffardi, O.; Pantaleoni, C.; Binelli, S. Seizures and EEG features in 74 Patients With Genetic Dysmorphic Syndromes. Am. J. Med. Genet. A 2014, 164A, 3154–3161. [Google Scholar] [CrossRef] [PubMed]
- Scocchia, A.; Wigby, K.M.; Masser-Frye, D.; Del Campo, M.; Galarreta, C.I.; Thorpe, E.; McEachern, J.; Robinson, K.; Gross, A.; Ajay, S.S.; et al. Clinical whole genome sequencing as a first-tier test at a resource-limited dysmorphology clinic in Mexico. NPJ Genom. Med. 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratt, P.W.E.; Ben-Shalom, R.; Keeshen, C.M.; Burke, K.J., Jr.; Clarkson, R.L.; Sanders, S.J.; Bender, K.J. The Autism-Associated Gene SCN2A Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 2019, 103, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE commission for classification and terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogliati, F.; Giorgini, V.; Masciadri, M.; Bonati, M.T.; Marchi, M.; Cracco, I.; Gentilini, D.; Peron, A.; Savini, M.N.; Spaccini, L.; et al. Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. Int. J. Mol. Sci. 2019, 24, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, A.; Enoki, H.; Niimi, K.; Nozaki, T.; Baba, S.; Shibamoto, I.; Otsuki, Y.; Oanishi, T. Epilepsy in patients with focal cortical dysplasia may be associated with autism spectrum disorder. Epilepsy Beav. 2021, 120, 107990. [Google Scholar] [CrossRef] [PubMed]
- Stosser, M.B.; Lindy, A.S.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.T.; Hollingsworth, G.; Muir, A.M.; Schneider, A.L.; Thuesmunn, Z.; Knupp, A.; King, C.; Lacroix, A.; Mehaffey, M.G.; Berkovic, S.F.; et al. Parental mosaicism in “de novo” epileptic encephalopathies. N. Engl. J. Med. 2018, 378, 1646–1648. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.M.; King, C.; Schneider, A.L.; Buttar, A.S.; Scheffer, I.E.; Sadleir, L.G.; Mefford, H.C. Double somatic mosaicism in a child with Dravet syndrome. Neurol. Genet. 2019, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Case | Clinical Picture | Onset of Neurological and Neuropsychological Impairment | Epilepsy Onset | Circadian Seizure Incidence | EEG | Effective AEDS |
|---|---|---|---|---|---|---|
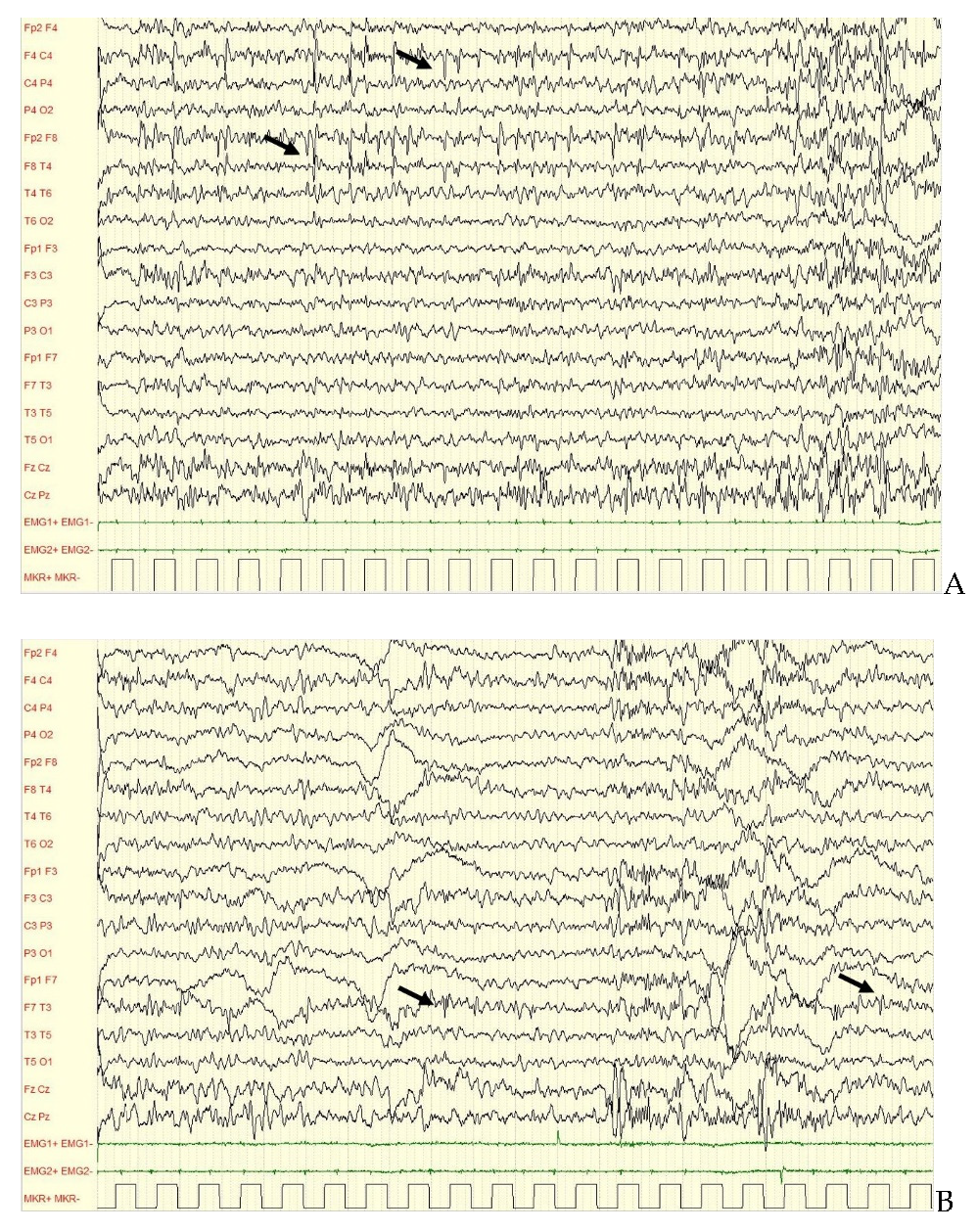
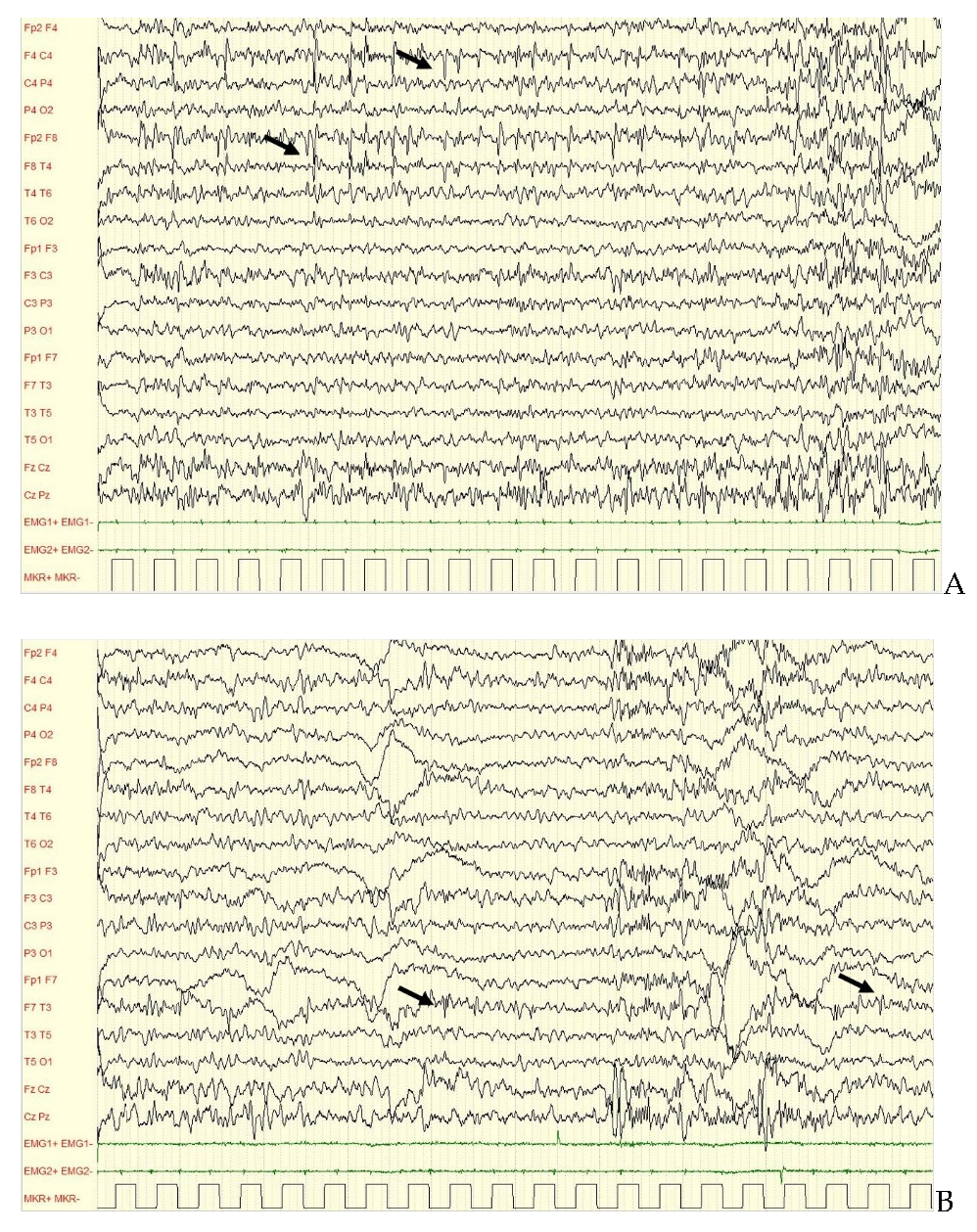
| 1 | EE Ohtahara-like, spastic tetraparesis, CVI, severe ID. | First year of life | Second day of life (from 19 months: mainly focal seizures) | Both during wakefulness and sleep | Initially BSP, then SBA, EA predominant over temporal areas. | TPM, VPA |
| 2 | Early Myoclonic Encephalopathy, spastic tetraparesis, severe ID. | First year of life | Second day of life | Both during wakefulness and sleep | Initially BSP, then POBA. Subcontinous SWA and EA predominant over occipital areas. | Drug resistance |
| 3 | ASD, ID—medium ID, focal epilepsy | First year of life | 2 years of life | Mainly during wakefulness | IBA, EA over frontotemporal areas bilaterally. | VPA |
| 4 | ASD, severe ID, focal epilepsy | First year of life | 8 years of life | Rare during wakefulness, mainly during sleep. | NBA, SA over right frontal areas. EA over right temporal areas. | Initially VPA, then CBZ (after 14 years of life) |
| 5 | Medium ID, focal epilepsy (late onset) | Second year of life | 26 years of life | During sleep | NBA, SA and EA predominant over right frontal areas. | CBZ + CLB |
| Case | Clinical | Mutation Type | Mutation | Origin |
|---|---|---|---|---|
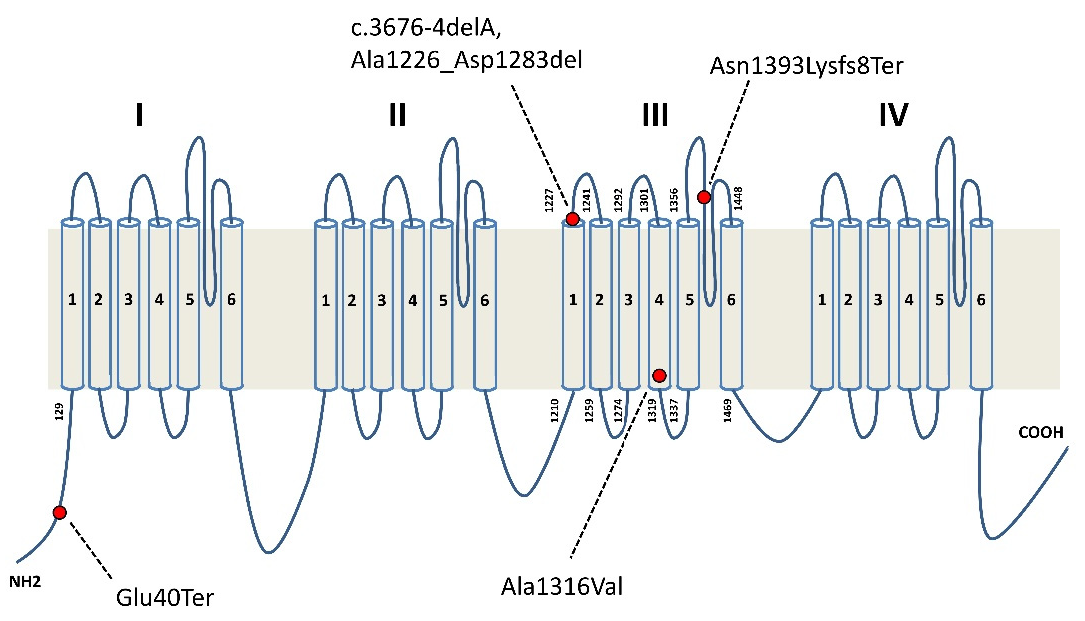
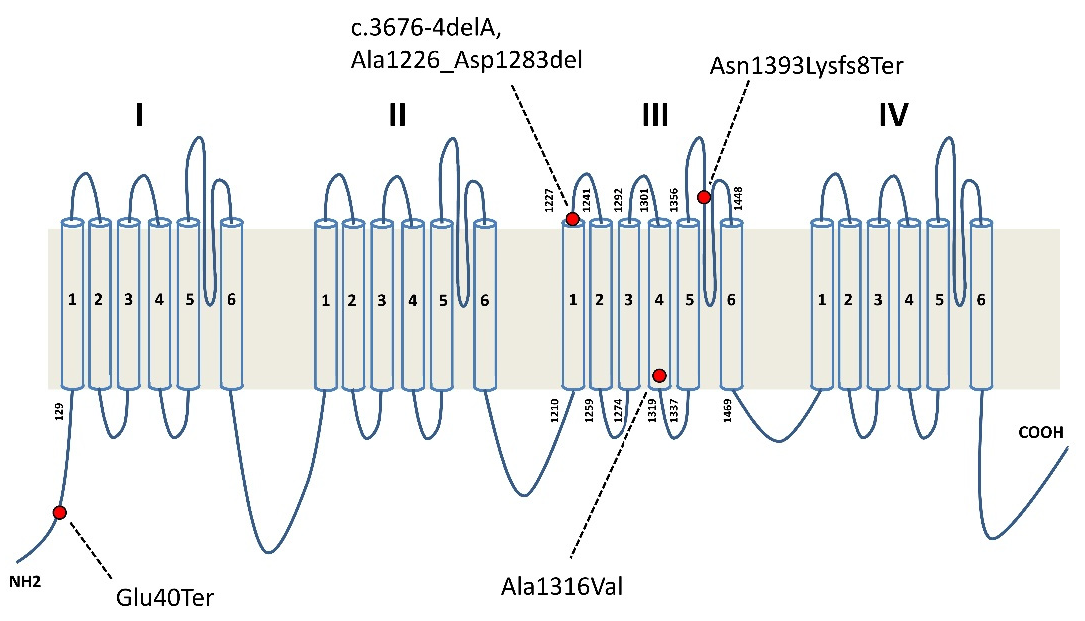
| 1 | Typical EIEE related to SCN2A mutation | Missense | p.Ala1316Val | De novo |
| 2 | Typical EIEE related to SCN2A mutation | Missense | p.Ala1316Val | De novo |
| 3 | ASD, low IQ, focal epilepsy | Splice site | p.Ala1226_Asp1283del | De novo |
| 4 | ASD, low IQ, focal epilepsy | Frameshift | c.4176-4179delCAAT; p.Asn1393LysfsTer8 | De novo |
| 5 | Low IQ, late onset focal epilepsy | Nonsense | p.Glu40Ter | From mosaic father |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Epifanio, R.; Giorda, R.; Merlano, M.C.; Zanotta, N.; Romaniello, R.; Marelli, S.; Russo, S.; Cogliati, F.; Bassi, M.T.; Zucca, C. SCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features. Brain Sci. 2022, 12, 18. https://doi.org/10.3390/brainsci12010018
Epifanio R, Giorda R, Merlano MC, Zanotta N, Romaniello R, Marelli S, Russo S, Cogliati F, Bassi MT, Zucca C. SCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features. Brain Sciences. 2022; 12(1):18. https://doi.org/10.3390/brainsci12010018
Chicago/Turabian StyleEpifanio, Roberta, Roberto Giorda, Maria Carolina Merlano, Nicoletta Zanotta, Romina Romaniello, Susan Marelli, Silvia Russo, Francesca Cogliati, Maria Teresa Bassi, and Claudio Zucca. 2022. "SCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features" Brain Sciences 12, no. 1: 18. https://doi.org/10.3390/brainsci12010018
APA StyleEpifanio, R., Giorda, R., Merlano, M. C., Zanotta, N., Romaniello, R., Marelli, S., Russo, S., Cogliati, F., Bassi, M. T., & Zucca, C. (2022). SCN2A Pathogenic Variants and Epilepsy: Heterogeneous Clinical, Genetic and Diagnostic Features. Brain Sciences, 12(1), 18. https://doi.org/10.3390/brainsci12010018