
Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample
2.2. Assessment of Bulbar Function
2.3. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Kolb, S.J.; Battle, D.J.; Dreyfuss, G. Molecular functions of the SMN complex. J. Child Neurol. 2007, 22, 990–994. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Willig, T.; Paulus, J.; Saintguily, J.L.; Béon, C.; Navarro, J. Swallowing problems in neuromuscular disorders. Arch. Phys. Med. Rehabil. 1994, 75, 1175–1181. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; Wijngaarde, C.A.; Stam, M.; Bartels, B.; Otto, L.A.; Lemmink, H.H.; Schoenmakers, M.A.; Cuppen, I.; Berg, L.H.V.D.; Van Der Pol, W.L. Muscle strength and motor function throughout life in a cross-sectional cohort of 180 patients with spinal muscular atrophy types 1c–4. Eur. J. Neurol. 2018, 25, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Darras, B.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Hagenacker, T.; Wurster, C.D.; Günther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef]
- Walter, M.C.; Wenninger, S.; Thiele, S.; Stauber, J.; Hiebeler, M.; Greckl, E.; Stahl, K.; Pechmann, A.; Lochmüller, H.; Kirschner, J.; et al. Safety and treatment effects of Nusinersen in longstanding adult 5q-SMA type 3—A prospective observational study. J. Neuromuscul. Dis. 2019, 6, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggi, L.; Bello, L.; Bonanno, S.; Govoni, A.; Caponnetto, C.; Passamano, L.; Grandis, M.; Trojsi, F.; Cerri, F.; Ferraro, M.; et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1166–1174. [Google Scholar] [CrossRef]
- Van den Engel-Hoek, L.; Erasmus, C.E.; Van Bruggen, H.W.; De Swart, B.J.M.; Sie, L.T.L.; Steenks, M.H.; De Groot, I.J.M. Dysphagia in spinal muscular atrophy type II: More than a bulbar problem? Neurology 2009, 73, 1787–1791. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; van Bruggen, H.W.; Witkamp, T.D.; Sparreboom-Kalaykova, S.I.; Stam, M.; Berg, L.H.V.D.; Steenks, M.H.; van der Pol, W.L. Bulbar muscle MRI changes in patients with SMA with reduced mouth opening and dysphagia. Neurology 2014, 83, 1060–1066. [Google Scholar] [CrossRef]
- Van Bruggen, H.W.; Wadman, R.I.; Bronkhorst, E.M.; Leeuw, M.; Creugers, N.; Kalaykova, S.I.; Steenks, M.H. Mandibular dysfunction as a reflection of bulbar involvement in SMA type 2 and 3. Neurology 2016, 86, 552–559. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Product Information Spinraza. 2017. Available online: https://www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_en.pdf (accessed on 15 May 2021).
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Bohlender, J.E.; Frick, S.; Colotto, U.; Hotzenköcherle, S.; Brockmann-Bauser, M. Der deutsche Sydney Swallow Questionnaire. Reliabilität und Validität bei Patienten mit oropharyngealer Dysphagie. HNO 2021. [Google Scholar] [CrossRef]
- O’Hagen, J.M.; Glanzman, A.M.; McDermott, M.P.; Ryan, P.A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W.B.; et al. An expanded version of the hammersmith functional motor scale for SMA II and III patients. Neuromuscul. Disord. 2007, 17, 693–697. [Google Scholar] [CrossRef]
- Mazzone, E.S.; Mayhew, A.; Montes, J.; Ramsey, D.; Fanelli, L.; Young, S.D.; Mercuri, E. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve 2017, 55, 869–874. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Forcina, N.; Mazzone, E.S.; Scoto, M.; Montes, J.; Pasternak, A.; Mayhew, A.; Messina, S.; Sframeli, M.; et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017, 17, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrattan, K.E.; Graham, R.J.; DiDonato, C.J.; Darras, B.T. Dysphagia phenotypes in spinal muscular atrophy: The past, present, and promise for the future. Am. J. Speech-Lang. Pathol. 2021, 30, 1008–1022. [Google Scholar] [CrossRef]
- Wijngaarde, C.A.; Stam, M.; Otto, L.A.; Bartels, B.; Asselman, F.-L.; van Eijk, R.P.; Berg, L.H.V.D.; Goedee, H.S.; Wadman, R.I.; van der Pol, W.L. Muscle strength and motor function in adolescents and adults with spinal muscular atrophy. Neurology 2020, 95, e1988–e1998. [Google Scholar] [CrossRef]
- Kaufmann, P.; McDermott, M.P.; Darras, B.; Finkel, R.S.; Sproule, D.M.; Kang, P.; Oskoui, M.; Constantinescu, A.; Gooch, C.; Foley, A.R.; et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 1889–1897. [Google Scholar] [CrossRef]
- Kruse, T.; Heller, R.; Wirth, B.; Glöggler, J.; Wurster, C.D.; Ludolph, A.C.; Braumann, B. Maximum bite force in patients with spinal muscular atrophy during the first year of nusinersen therapy—A pilot study. Acta Myol. 2020, 39, 83–89. [Google Scholar]
- Duong, T.; Wolford, C.; McDermott, M.P.; Macpherson, C.E.; Pasternak, A.; Glanzman, A.M.; Martens, W.B.; Kichula, E.; Darras, B.T.; De Vivo, D.C.; et al. Nusinersen treatment in adults with spinal muscular atrophy. Neurol. Clin. Pr. 2021, 11, e317–e327. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in type 1 spinal muscular atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Dixon, S.; Naik, R.; Noone, J.M.; Ms, J.D.B.; Whitmire, S.M.; Ba, R.M.; Arnold, W. Early experiences of nusinersen for the treatment of spinal muscular atrophy: Results from a large survey of patients and caregivers. Muscle Nerve 2020, 63, 311–319. [Google Scholar] [CrossRef]
- Kraus, E.-M.; Rommel, N.; Stoll, L.H.; Oettinger, A.; Vogel, A.P.; Synofzik, M. Validation and psychometric properties of the German version of the SWAL-QOL. Dysphagia 2018, 33, 431–440. [Google Scholar] [CrossRef]
- Bauer, F.; Seiss, M.; Gräßel, E.; Stelzle, F.; Klotz, M.; Rosanowski, F. Schluckbezogene Lebensqualität bei Mundhöhlenkarzinomen Anderson-dysphagia-inventory, Deutsche version. HNO 2010, 58, 692–697. [Google Scholar] [CrossRef]
- Zaretsky, E.; Steinbach-Hundt, S.; Pluschinski, P.; Grethel, I.; Hey, C. Validation of the German version of eating assessment tool for head and neck cancer patients. Laryngo-Rhino-Otologie 2018, 97, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Audag, N.; Liistro, G.; Goubau, C.; Vandervelde, L.; Poncin, W.; Toussaint, M.; Reychler, G. Screening for oropharyngeal dysphagia in adult patients with neuromuscular diseases using the Sydney Swallow Questionnaire. Muscle Nerve 2021, 64, 277–284. [Google Scholar] [CrossRef]
- Elsheikh, B.; Prior, T.; Zhang, X.; Miller, R.; Kolb, S.J.; Moore, D.; Bradley, W.; Barohn, R.; Bryan, W.; Gelinas, D.; et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve 2009, 40, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.; Burke, T.; Vajda, A.; Heverin, M.; Hardiman, O. What does the ALSFRS-R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 88, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Chiriboga, C.A.; Iannaccone, S.T.; Swoboda, K.J.; Montes, J.; Mignon, L.; Darryl, C. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology. 2019, 92, e2492–e2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Baranello, G.; Kirschner, J.; Servais, L.; Goemans, N.; Pera, M.C.; Khwaja, O. SUNFISH Part 1: Risdiplam (RG7916) treatment results in a sustained increase of SMN protein levels and improvement in motor function in patients with Type 2 or 3 SMA. In Proceedings of the 23rd International Annual Congress of the World Muscle Society, Mendoza, Argentina, 2–6 October 2018. [Google Scholar]
- Olthoff, A.; Carstens, P.-O.; Zhang, S.; von Fintel, E.; Friede, T.; Lotz, J.; Frahm, J.; Schmidt, J. Evaluation of dysphagia by novel real-time MRI. Neurology 2016, 87, 2132–2138. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sex | Male (%) | Female (%) | ||
|---|---|---|---|---|
| 13 (59) | 9 (41) | |||
| Presence of Spondylodesis | Yes (%) | No (%) | ||
| 10 (45) | 12 (54) | |||
| SMA type | 2 (%) | 3 (%) | ||
| 12 (54) | 10 (45) | |||
| SMN 2 Copy Number | 3 (%) | 4 (%) | 5 (%) | ≥6 (%) |
| 14 (64) | 6 (27) | 1 (4.5) | 1 (4.5) | |
| Age | Mean (sd) | Range | Minimum | Maximum |
| 38.5 (14.2) | 52 | 20 | 72 | |
| Symptom Duration in Years | Mean (sd) | Range | Minimum | Maximum |
| 34.3 (11.9) | 44 | 14 | 58 |
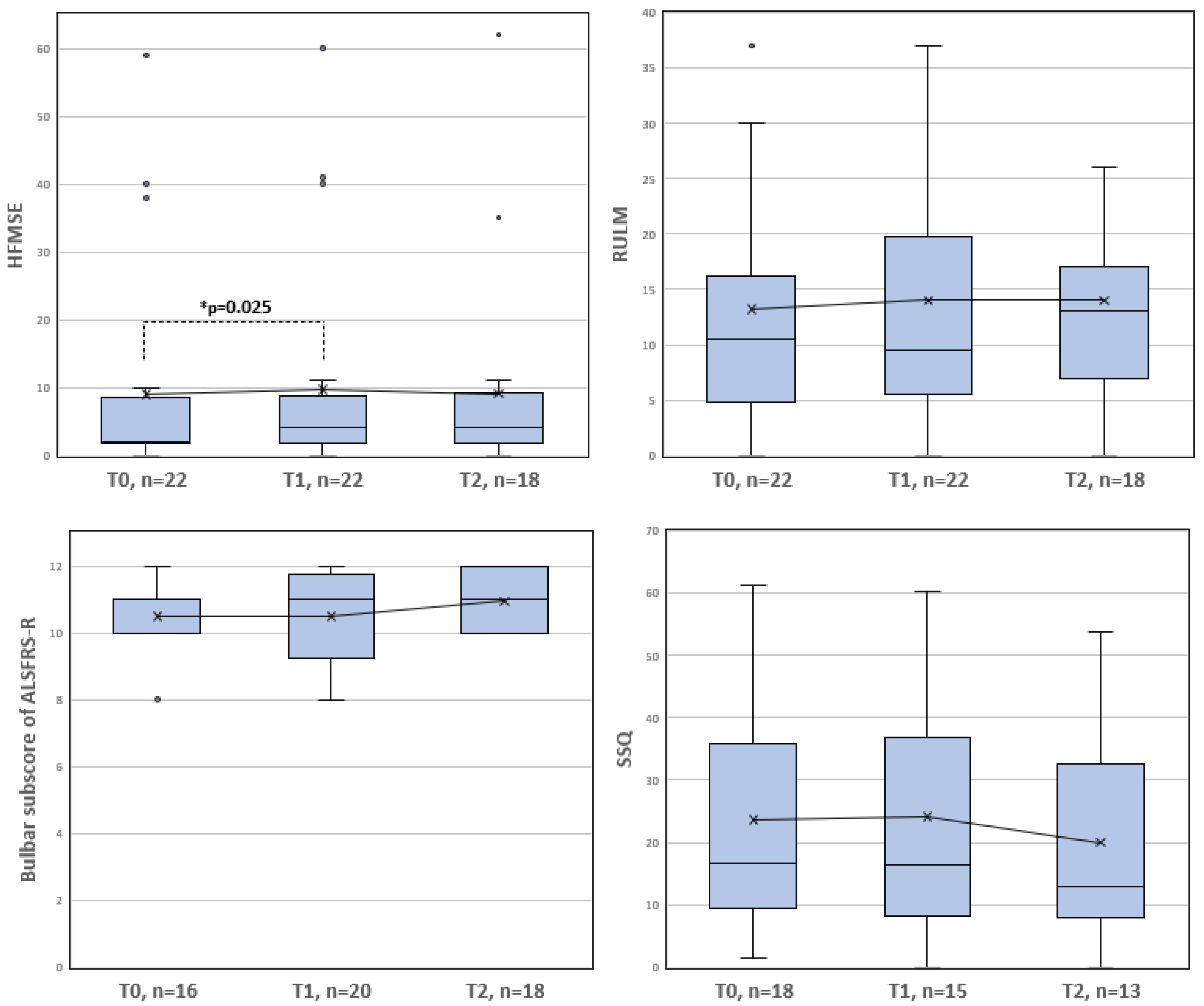
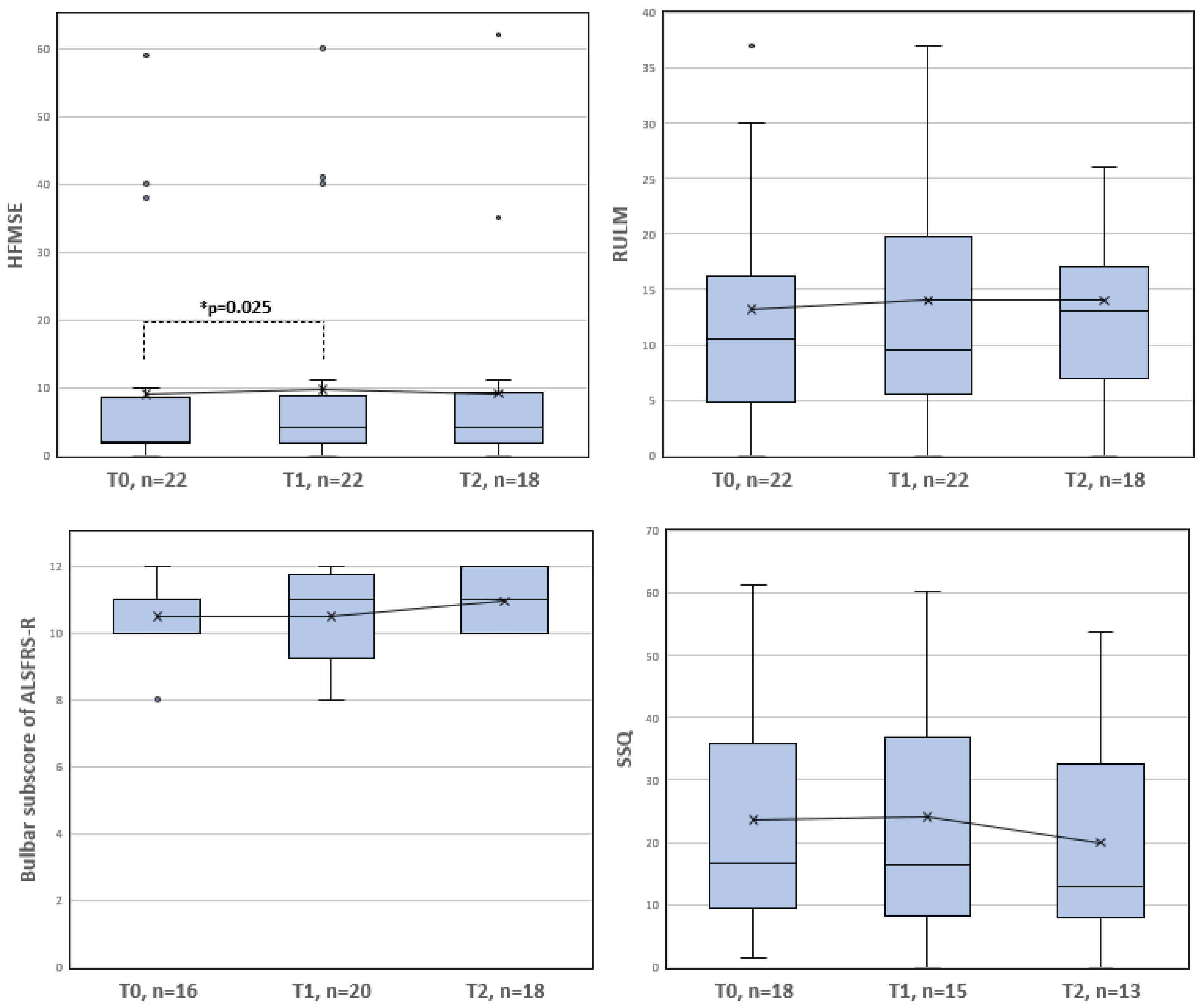
| N | Mean (sd) | Minimum | Maximum | Lower Quartile | Median | Upper Quartile | |
|---|---|---|---|---|---|---|---|
| Bulbar subscore of ALSFRS-R (0–12) | |||||||
| T0 | 16 | 10.50 (1.21) | 8.0 | 12.0 | 10.0 | 11.0 | 11.0 |
| T1 | 20 | 10.50 (1.32) | 8.0 | 12.0 | 9.25 | 11.0 | 11.75 |
| T2 | 18 | 10.94 (0.97) | 10.0 | 12.0 | 10.0 | 11.0 | 12.0 |
| SSQ (0–100) | |||||||
| T0 | 18 | 23.59 (18.85) | 1.47 | 61.18 | 9.48 | 16.69 | 35.95 |
| T1 | 15 | 24.12 (19.32) | 0.0 | 60.29 | 8.23 | 16.05 | 36.88 |
| T2 | 13 | 19.95 (17.86) | 0.0 | 53.82 | 8.00 | 12.81 | 32.48 |
| HFMSE (0–66) | |||||||
| T0 | 22 | 8.95 (15.64) | 0.0 | 59.0 | 1.75 | 2.0 | 8.5 |
| T1 | 22 | 9.68 (15.92) | 0.0 | 60.0 | 1.75 | 4.0 | 8.75 |
| T2 | 18 | 9.06 (15.46) | 0.0 | 62.0 | 1.75 | 4.0 | 9.25 |
| RULM (0–37) | |||||||
| T0 | 22 | 13.23 12.04) | 0.0 | 37.0 | 4.75 | 10.5 | 16.25 |
| T1 | 22 | 14.0 (12.11) | 0.0 | 37.0 | 5.5 | 9.5 | 19.75 |
| T2 | 18 | 14.0 (10.57) | 0.0 | 37.0 | 7.0 | 13.0 | 17.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brakemeier, S.; Stolte, B.; Thimm, A.; Kizina, K.; Totzeck, A.; Munoz-Rosales, J.; Kleinschnitz, C.; Hagenacker, T. Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen. Brain Sci. 2021, 11, 1244. https://doi.org/10.3390/brainsci11091244
Brakemeier S, Stolte B, Thimm A, Kizina K, Totzeck A, Munoz-Rosales J, Kleinschnitz C, Hagenacker T. Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen. Brain Sciences. 2021; 11(9):1244. https://doi.org/10.3390/brainsci11091244
Chicago/Turabian StyleBrakemeier, Svenja, Benjamin Stolte, Andreas Thimm, Kathrin Kizina, Andreas Totzeck, Juan Munoz-Rosales, Christoph Kleinschnitz, and Tim Hagenacker. 2021. "Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen" Brain Sciences 11, no. 9: 1244. https://doi.org/10.3390/brainsci11091244
APA StyleBrakemeier, S., Stolte, B., Thimm, A., Kizina, K., Totzeck, A., Munoz-Rosales, J., Kleinschnitz, C., & Hagenacker, T. (2021). Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen. Brain Sciences, 11(9), 1244. https://doi.org/10.3390/brainsci11091244