
Epigenetic Mediation of AKT1 rs1130233’s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing

 ,
,  ,
,
Abstract
:1. Introduction
2. Methods
2.1. Participants
2.2. Experimental Design
2.3. Image Acquisition
2.4. fMRI Task
2.5. Genotyping and Methylation Assay
2.6. Image Analysis
3. Results
3.1. Participants
3.2. Symptomatic and Behavioural Response to THC
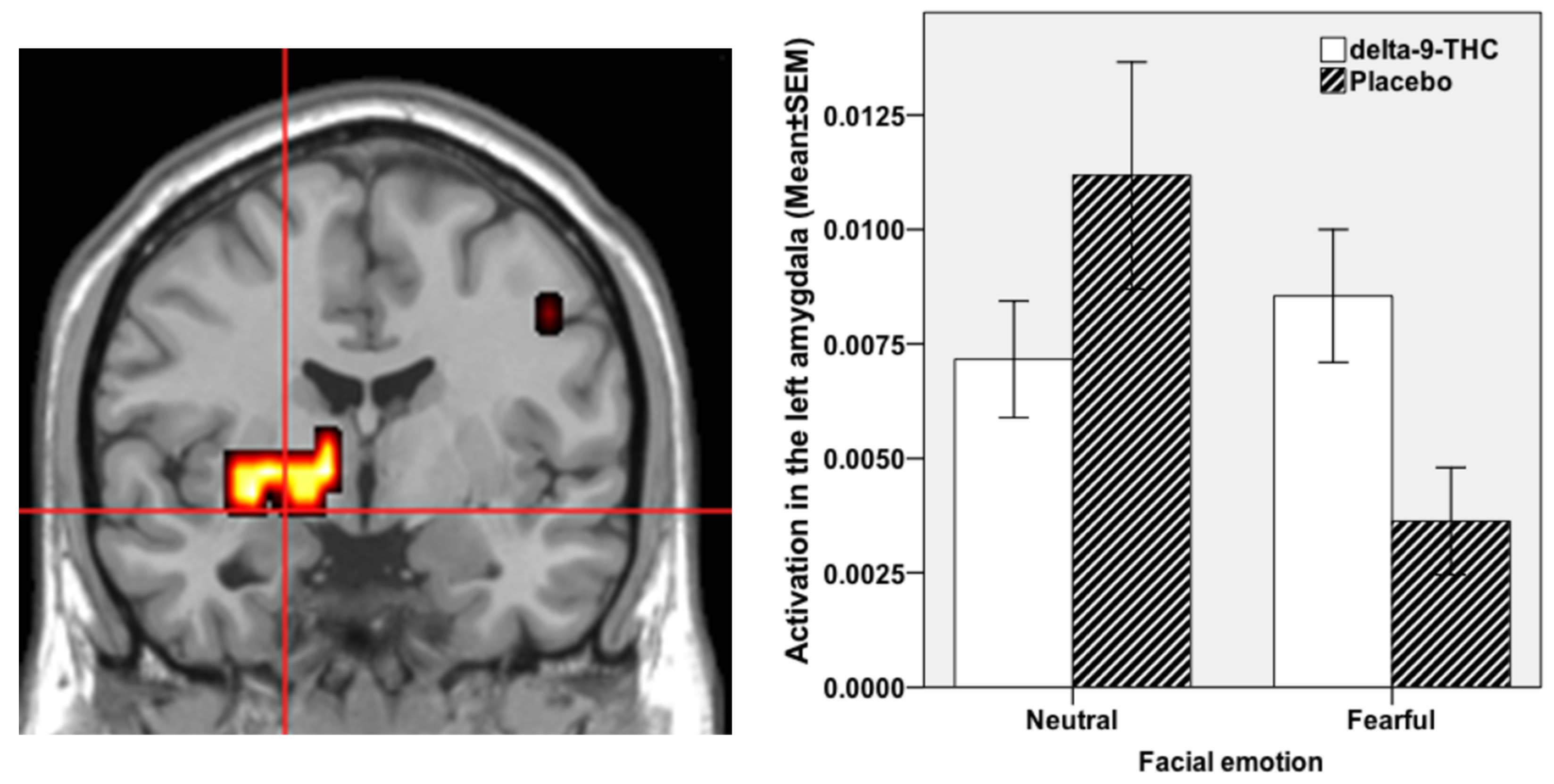
3.3. Regional Brain Response to THC during Fear Processing
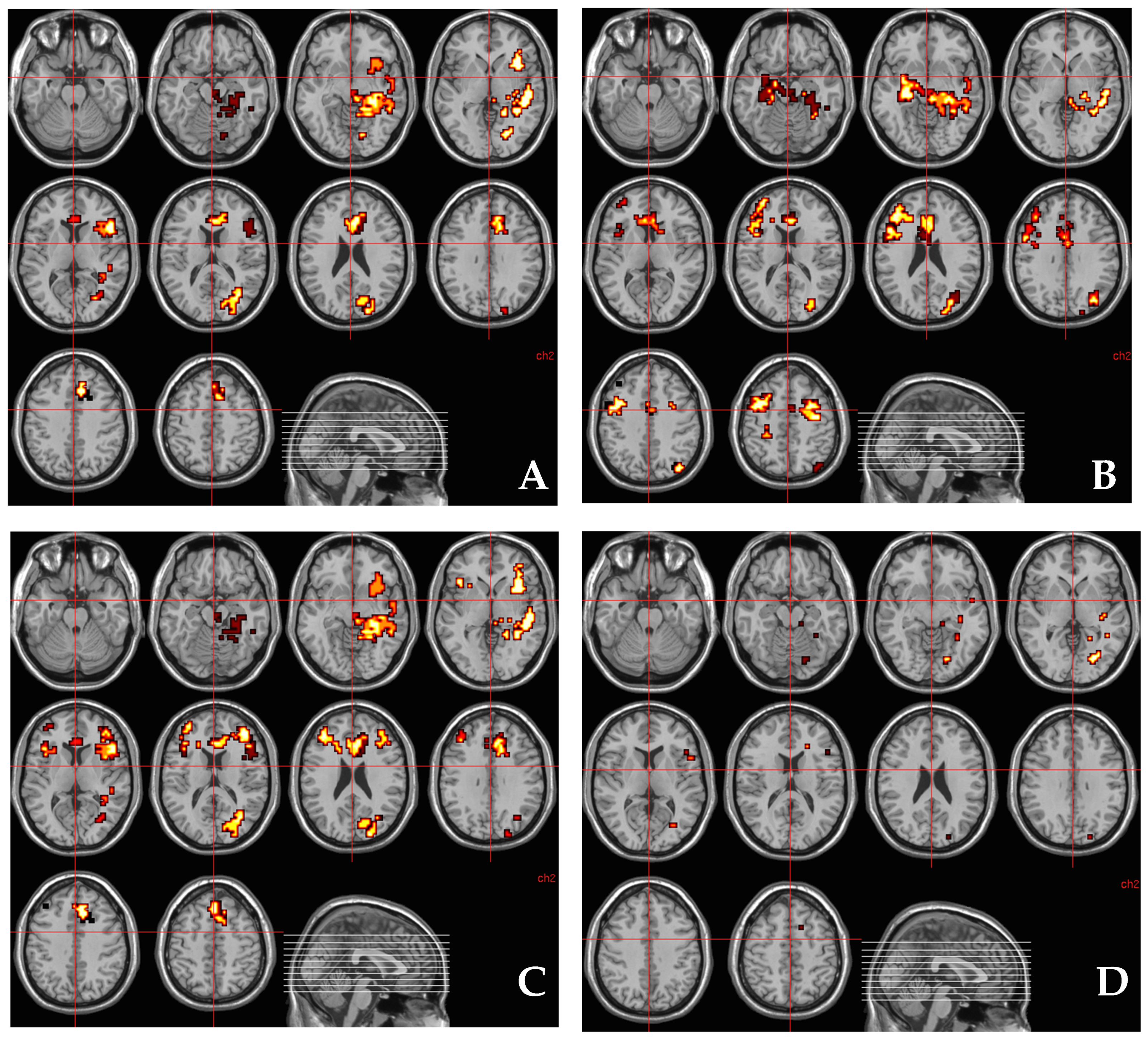
3.4. Relationship between AKT1 Genotype (rs1130233) and the Effect of THC on Regional Brain Activation during Fear Processing
3.5. Relationship between Methylation at CpG11–12 Site around AKT1 SNP (rs1130233) and the Effect of THC on Regional Brain Activation during Fear Processing
3.6. Relationship between Methylation at CpG11–12 Site around AKT1 SNP (rs1130233) and the Number of A Alleles at AKT1 SNP (rs1130233)
3.7. Relationship between AKT1 Genotype (rs1130233) and the Effect of THC on Regional Brain Activation during Fear Processing after Covarying for Methylation at CpG11–12 Site
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethical Standards
References
- UNODC. World Drug Report. Available online: https://wdr.unodc.org/wdr2019/ (accessed on 28 July 2021).
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Phytocannabinoids. Progress in the Chemistry of Organic Natural Products; Kinghorn, A., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Cham, Switzerland, 2017; Volume 103. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Munoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict. Biol. 2008, 13, 147–159. [Google Scholar] [CrossRef]
- Ashton, C.H. Pharmacology and effects of cannabis: A brief review. Br. J. Psychiatry 2001, 178, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, F.A.; Wotjak, C.T. Cannabinoids and Anxiety. Curr. Top. Behav. Neurosci. 2009, 2, 429–450. [Google Scholar] [CrossRef]
- Phan, K.L.; Angstadt, M.; Golden, J.; Onyewuenyi, I.; Popovska, A.; de Wit, H. Cannabinoid Modulation of Amygdala Reactivity to Social Signals of Threat in Humans. J. Neurosci. 2008, 28, 2313–2319. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; Crippa, J.; Kambeitz, J.; Malhi, S.; Giampietro, V.; Williams, S.; Brammer, M.; Rubia, K.; et al. Impairment of inhibitory control processing related to acute psychotomimetic effects of cannabis. Eur. Neuropsychopharmacol. 2015, 25, 26–37. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Egerton, A.; Kim, E.; Rosso, L.; Barros, D.R.; Hammers, A.; Brammer, M.; Turkheimer, F.; Howes, O.; McGuire, P. Acute induction of anxiety in humans by delta-9-tetrahydrocannabinol related to amygdalar cannabinoid-1 (CB1) receptors. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Fusar-Poli, P.; Borgwardt, S.; Martin-Santos, R.; Nosarti, C.; O’Carroll, C.; Allen, P.; Seal, M.; Fletcher, P.C.; Crippa, J.A.; et al. Modulation of Mediotemporal and Ventrostriatal Function in Humans by Δ9-Tetrahydrocannabinol. Arch. Gen. Psychiatry 2009, 66, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Colizzi, M.; McGuire, P.; Giampietro, V.; Williams, S.; Brammer, M.; Bhattacharyya, S. Previous cannabis exposure modu-lates the acute effects of delta-9-tetrahydrocannabinol on attentional salience and fear processing. Exp. Clin. Psychopharmacol. 2018, 26, 582. [Google Scholar] [CrossRef]
- Boggs, D.L.; Nguyen, J.; Morgenson, D.; A Taffe, M.; Ranganathan, M. Clinical and Preclinical Evidence for Functional Interactions of Cannabidiol and Δ9-Tetrahydrocannabinol. Neuropsychopharmacology 2018, 43, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Morrison, P.D.; Fusar-Poli, P.; Martin-Santos, R.; Borgwardt, S.; Wintonbrown, T.T.; Nosarti, C.; Carroll, C.M.O.; Seal, M.L.; Allen, P.; et al. Opposite Effects of Δ-9-Tetrahydrocannabinol and Cannabidiol on Human Brain Function and Psychopathology. Neuropsychopharmacology 2010, 35, 764–774. [Google Scholar] [CrossRef]
- Crippa, J.A.S.; Derenusson, G.N.; Ferrari, T.B.; Wichert-Ana, L.; Duran, F.L.; Martin-Santos, R.; Simões, M.V.; Bhattacharyya, S.; Fusar-Poli, P.; Atakan, Z.; et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: A preliminary report. J. Psychopharmacol. 2011, 25, 121–130. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Crippa, J.A.; Allen, P.; Martin-Santos, R.; Borgwardt, S.; Fusar-Poli, P.; Rubia, K.; Kambeitz, J.; O’Carroll, C.; Seal, M.; et al. Induction of Psychosis byΔ9-Tetrahydrocannabinol Reflects Modulation of Prefrontal and Striatal Function During Attentional Salience Processing. Arch. Gen. Psychiatry 2012, 69, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Falkenberg, I.; Martin-Santos, R.; Atakan, Z.; A Crippa, J.; Giampietro, V.; Brammer, M.; McGuire, P. Cannabinoid Modulation of Functional Connectivity within Regions Processing Attentional Salience. Neuropsychopharmacology 2014, 40, 1343–1352. [Google Scholar] [CrossRef]
- Colizzi, M.; Bhattacharyya, S. Does Cannabis Composition Matter? Differential Effects of Delta-9-tetrahydrocannabinol and Cannabidiol on Human Cognition. Curr. Addict. Rep. 2017, 4, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Iyegbe, C.; Atakan, Z.; Martin-Santos, R.; Crippa, J.A.; Xu, X.; Williams, S.; Brammer, M.; Rubia, K.; Prata, D.; et al. Protein kinase B (AKT1) genotype mediates sensitivity to cannabis-induced impairments in psychomotor control. Psychol. Med. 2014, 44, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Van Winkel, R. Family-Based Analysis of Genetic Variation Underlying Psychosis-Inducing Effects of CannabisSibling Analysis and Proband Follow-upGenetic Variation Underlying Cannabis Effects. Arch. Gen. Psychiatry 2011, 68, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Blasi, G.; Napolitano, F.; Ursini, G.; Taurisano, P.; Romano, R.; Caforio, G.; Fazio, L.; Gelao, B.; Di Giorgio, A.; Iacovelli, L.; et al. DRD2/AKT1 interaction on D2 c-AMP independent signaling, attentional processing, and response to olanzapine treatment in schizophrenia. Proc. Natl. Acad. Sci. USA 2011, 108, 1158–1163. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, E.; Zucali, P.A.; Peters, G.J.; Cortesi, F.; D’Incecco, A.; Smit, E.F.; Falcone, A.; Burgers, J.A.; Santoro, A.; Danesi, R.; et al. Association of Polymorphisms in AKT1 and EGFR with Clinical Outcome and Toxicity in Non–Small Cell Lung Cancer Patients Treated with Gefitinib. Mol. Cancer Ther. 2010, 9, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Gil, G.; Robins, H.; Hu, W.; Hirshfield, K.; Bond, E.; Bond, G.; Levine, A.J. Detection of functional single-nucleotide polymorphisms that affect apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 16297–16302. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.-Y.; Nicodemus, K.K.; Chen, Q.; Li, Z.; Brooke, J.K.; Honea, R.; Kolachana, B.S.; Straub, R.E.; Meyer-Lindenberg, A.; Sei, Y.; et al. Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J. Clin. Investig. 2008, 118, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Gainetdinov, R.; Caron, M.G. The Akt–GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Dunleavy, M.; Henshall, D.C. Cell Signaling Underlying Epileptic Behavior. Front. Behav. Neurosci. 2011, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, S.; Ikeda, Y.; Murakami, M.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y. Roles of PI3K/AKT/GSK3 Pathway Involved in Psychiatric Illnesses. Diseases 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.; Gai, H.; Su, R.; Deji, C.; Cui, J.; Lai, J.; Zhu, Y. PI3K-AKT-GSK3β-CREB signaling pathway regulates anxiety-like behavior in rats following alcohol withdrawal. J. Affect. Disord. 2018, 235, 96–104. [Google Scholar] [CrossRef]
- Bossong, M.G.; Van Berckel, B.N.M.; Boellaard, R.; Zuurman, L.; Schuit, R.C.; Windhorst, A.; A Van Gerven, J.M.; Ramsey, N.; Lammertsma, A.; Kahn, R.S. Δ9-Tetrahydrocannabinol Induces Dopamine Release in the Human Striatum. Neuropsychopharmacology 2008, 34, 759–766. [Google Scholar] [CrossRef]
- Sami, M.; Rabiner, E.A.; Bhattacharyya, S. Does cannabis affect dopaminergic signaling in the human brain? A systematic review of evidence to date. Eur. Neuropsychopharmacol. 2015, 25, 1201–1224. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Crippa, J.A.; Martin-Santos, R.; Winton-Brown, T.; Fusar-Poli, P. Imaging the neural effects of canna-binoids: Current status and future opportunities for psychopharmacology. Curr. Pharm. Des. 2009, 15, 2603–2614. [Google Scholar] [CrossRef]
- Ozaita, A.; Puighermanal, E.; Maldonado, R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J. Neurochem. 2007, 102, 1105–1114. [Google Scholar] [CrossRef]
- Shum, C.; Dutan, L.; Annuario, E.; Cornish, K.W.; Taylor, S.E.; Taylor, R.D.; Andreae, L.; Buckley, N.J.; Price, J.; Bhattacharyya, S.; et al. Δ9-tetrahydrocannabinol and 2-AG decreases neurite outgrowth and differentially affects ERK1/2 and Akt signaling in hiPSC-derived cortical neurons. Mol. Cell. Neurosci. 2020, 103, 103463. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; A Crippa, J.; Kambeitz, J.; Prata, D.; Williams, S.; Brammer, M.; A Collier, D.; McGuire, P. Preliminary report of biological basis of sensitivity to the effects of cannabis on psychosis: AKT1 and DAT1 genotype modulates the effects of δ-9-tetrahydrocannabinol on midbrain and striatal function. Mol. Psychiatry 2012, 17, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, A.T.; Luborsky, L.; E Woody, G.; O’Brien, C.P. An Improved Diagnostic Evaluation Instrument for Substance Abuse Patients. J. Nerv. Ment. Dis. 1980, 168, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kay, S.R.; Fiszbein, A.; Opler, L.A. The Positive and Negative Syndrome Scale (PANSS) for Schizophrenia. Schizophr. Bull. 1987, 13, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Spielberger, C. Manual of the State-Trait Anxiety Inventory; Consulting Psychologists Press Inc.: Palo Alto, CA, USA, 1983. [Google Scholar]
- Mathew, R.J.; Wilson, W.H.; Humphreys, D.F.; Lowe, J.V.; Wiethe, K.E. Regional Cerebral Blood Flow after Marijuana Smoking. Br. J. Pharmacol. 1992, 12, 750–758. [Google Scholar] [CrossRef]
- Norris, H. The action of sedatives on brain stem oculomotor systems in man. Neuropharmacology 1971, 10, 181–191. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Crippa, J.A.; Bhattacharyya, S.; Borgwardt, S.; Allen, P.; Martin-Santos, R.; Seal, M.; Surguladze, S.A.; O’Carrol, C.; Atakan, Z.; et al. Distinct Effects of Δ9-Tetrahydrocannabinol and Cannabidiol on Neural Activation During Emotional Processing. Arch. Gen. Psychiatry 2009, 66, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.; Smith, N.; Curtis, C.; Huckett, L.; Mill, J.; Craig, I.W. DNA from buccal swabs recruited by mail: Evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav. Genet. 2003, 33, 67–72. [Google Scholar] [CrossRef]
- Ouellet-Morin, I.; Wong, C.; Danese, A.; Pariante, C.M.; Papadopoulos, A.S.; Mill, J.; Arseneault, L. Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: A longitudinal study of discordant monozygotic twins. Psychol. Med. 2013, 43, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Coolen, M.W.; Statham, A.L.; Gardiner-Garden, M.; Clark, S.J. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: Critical evaluation and improvements. Nucleic Acids Res. 2007, 35, e119. [Google Scholar] [CrossRef] [Green Version]
- Brammer, M.; Bullmore, E.; Simmons, A.; Williams, S.; Grasby, P.; Howard, R.; Woodruff, P.; Rabe-Hesketh, S. Generic brain activation mapping in functional magnetic resonance imaging: A nonparametric approach. Magn. Reson. Imaging 1997, 15, 763–770. [Google Scholar] [CrossRef]
- Thirion, B.; Pinel, P.; Mériaux, S.; Roche, A.; Dehaene, S.; Poline, J.-B. Analysis of a large fMRI cohort: Statistical and methodological issues for group analyses. NeuroImage 2007, 35, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Hayasaka, S.; Nichols, T.E. Validating cluster size inference: Random field and permutation methods. Neuroimage 2003, 20, 2343–2356. [Google Scholar] [CrossRef]
- Bullmore, E.T.; Brammer, M.J.; Rabe-Hesketh, S.; Curtis, V.A.; Morris, R.G.; Williams, S.C.; McGuire, P.K. Methods for diagnosis and treatment of stimulus-correlated motion in generic brain activation studies using fMRI. Hum. Brain Mapp. 1999, 7, 38–48. [Google Scholar] [CrossRef]
- Friman, O.; Borga, M.; Lundberg, P.; Knutsson, H. Adaptive analysis of fMRI data. Neuroimage 2003, 19, 837–845. [Google Scholar] [CrossRef]
- Bullmore, E.; Long, C.; Suckling, J.; Fadili, J.; Calvert, G.; Zelaya, F.; Carpenter, T.A.; Brammer, M. Colored noise and computational inference in neurophysiological (fMRI) time series analysis: Resampling methods in time and wavelet domains. Hum. Brain Mapp. 2001, 12, 61–78. [Google Scholar] [CrossRef] [Green Version]
- Talairach, J.; Tournoux, P. Co-Planar Stereotaxic Atlas of the Human Brain; Thieme Medical: New York, NY, USA, 1988. [Google Scholar]
- Bullmore, E.; Suckling, J.; Overmeyer, S.; Rabe-Hesketh, S.; Taylor, E.; Brammer, M. Global, voxel, and cluster tests, by theory and permutation, for a difference between two groups of structural MR images of the brain. IEEE Trans. Med. Imaging 1999, 18, 32–42. [Google Scholar] [CrossRef]
- Baron, R.M.; Kenny, D.A. The moderator-mediator variable distinction in social psychological research: Conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 1986, 51, 1173–1182. [Google Scholar] [CrossRef]
- Fardo, D.W.; Becker, K.D.; Bertram, L.; Tanzi, R.E.; Lange, C. Recovering unused information in genome-wide association studies: The benefit of analyzing SNPs out of Hardy-Weinberg equilibrium. Eur. J. Hum. Genet. 2009, 17, 1676–1682. [Google Scholar] [CrossRef]
- Lai, W.-S.; Xu, B.; Westphal, K.G.C.; Paterlini, M.; Olivier, B.; Pavlidis, P.; Karayiorgou, M.; Gogos, J.A. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc. Natl. Acad. Sci. USA 2006, 103, 16906–16911. [Google Scholar] [CrossRef] [Green Version]
- Morgan, C.J.; Freeman, T.; Powell, J.; Curran, H.V. AKT1 genotype moderates the acute psychotomimetic effects of naturalistically smoked cannabis in young cannabis smokers. Transl. Psychiatry 2016, 6, e738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindocha, C.; Quattrone, D.; Freeman, T.; Murray, R.M.; Mondelli, V.; Breen, G.; Curtis, C.; Morgan, C.J.A.; Curran, H.V.; Di Forti, M. Do AKT1, COMT and FAAH influence reports of acute cannabis intoxication experiences in patients with first episode psychosis, controls and young adult cannabis users? Transl. Psychiatry 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Szutorisz, H.; Hurd, Y.L. Epigenetic Effects of Cannabis Exposure. Biol. Psychiatry 2016, 79, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Van Der Knaap, L.; Schaefer, J.M.; Franken, I.; Verhulst, F.; Van Oort, F.; Riese, H. Catechol-O-methyltransferase gene methylation and substance use in adolescents: The TRAILS study. Genes Brain Behav. 2014, 13, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9 -tetrahydrocannabinol, cannabidiol and Δ9 -tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batalla, A.; Crippa, J.; Busatto, G.; Guimaraes, F.; Zuardi, A.; Valverde, O.; Atakan, Z.; McGuire, P.; Bhattacharyya, S.; Martín-Santos, R. Neuroimaging Studies of Acute Effects of THC and CBD in Humans and Animals: A Systematic Review. Curr. Pharm. Des. 2014, 20, 2168–2185. [Google Scholar] [CrossRef]
- Bossong, M.; Jager, G.; Bhattacharyya, S.; Allen, P. Acute and Non-acute Effects of Cannabis on Human Memory Function: A Critical Review of Neuroimaging Studies. Curr. Pharm. Des. 2014, 20, 2114–2125. [Google Scholar] [CrossRef]
- Emamian, E.S.; Hall, D.; Birnbaum, M.; Karayiorgou, M.; A Gogos, J. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef]
- Bloomfield, M.; Ashok, A.; Volkow, N.D.; Howes, O.D. The effects of Δ9-tetrahydrocannabinol on the dopamine system. Nat. Cell Biol. 2016, 539, 369–377. [Google Scholar] [CrossRef]
- Colizzi, M.; Bhattacharyya, S. Neurocognitive effects of cannabis: Lessons learned from human experimental studies. Prog. Brain Res. 2018, 242, 179–216. [Google Scholar] [CrossRef]
- Colizzi, M.; McGuire, P.; Pertwee, R.; Bhattacharyya, S. Effect of cannabis on glutamate signalling in the brain: A systematic review of human and animal evidence. Neurosci. Biobehav. Rev. 2016, 64, 359–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colizzi, M.; Weltens, N.; McGuire, P.; Lythgoe, D.; Williams, S.; Van Oudenhove, L.; Bhattacharyya, S. Delta-9-tetrahydrocannabinol increases striatal glutamate levels in healthy individuals: Implications for psychosis. Mol. Psychiatry 2020, 25, 3231–3240. [Google Scholar] [CrossRef]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Tirotta, E.; Sotnikova, T.D.; Masri, B.; Salahpour, A.; Gainetdinov, R.R.; Borrelli, E.; Caron, M.G. Regulation of Akt Signaling by D2 and D3 Dopamine Receptors In Vivo. J. Neurosci. 2007, 27, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.G.; Ruiz-Llorente, L.; Sánchez, A.M.; Díaz-Laviada, I. Activation of phosphoinositide 3-kinase/PKB pathway by CB(1) and CB(2) cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF in-duction. Cell Signal 2003, 15, 851–859. [Google Scholar] [CrossRef]
- Wong, D.F.; Wagner, H.N.; Tune, L.E.; Dannals, R.F.; Pearlson, G.D.; Links, J.M.; Gjedde, A. Positron emission to-mography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science 1986, 234, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Arguello, P.A.; Gogos, J.A. A signaling pathway AKTing up in schizophrenia. J. Clin. Investig. 2008, 118, 2018–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Forti, M.; Iyegbe, C.; Sallis, H.; Kolliakou, A.; Falcone, M.A.; Paparelli, A.; Sirianni, M.; La Cascia, C.; Stilo, S.A.; Marques, T.R.; et al. Confirmation that the AKT1 (rs2494732) Genotype Influences the Risk of Psychosis in Cannabis Users. Biol. Psychiatry 2012, 72, 811–816. [Google Scholar] [CrossRef]
- Colizzi, M.; Iyegbe, C.; Powell, J.; Blasi, G.; Bertolino, A.; Murray, R.M.; Di Forti, M. Interaction between DRD2 and AKT1 genetic variations on risk of psychosis in cannabis users: A case–control study. NPJ Schizophr. 2015, 1, 15025. [Google Scholar] [CrossRef] [Green Version]
- Cortes, L.R.; Cisternas, C.D.; Forger, N.G. Does Gender Leave an Epigenetic Imprint on the Brain? Front. Neurosci. 2019, 13, 173. [Google Scholar] [CrossRef]
- Cuttler, C.; Mischley, L.K.; Sexton, M. Sex Differences in Cannabis Use and Effects: A Cross-Sectional Survey of Cannabis Users. Cannabis Cannabinoid Res. 2016, 1, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Lifetime Psychoactive Substance Use | |
|---|---|
| Caffeine | 33 subjects; Mean number of cups of coffee, tea or caffeinated drinks/day-2.42 (SD–1.86) (range 0–11) |
| Nicotine | 9 subjects; Mean number of cigarettes smoked/day-1.19 (SD–3.18) (range 0–15/day); 2 subjects smoked >10 cigarettes/day lifetime; only 1 subject smoked at that level at the time of the study. |
| Cannabis | <5 times used = 12 subjects; 5–25 times used = 24 subjects |
| Cocaine | A few times of reported use = 3 subjects |
| Amphetamines | A few times of reported use = 5 subjects Used small quantities from time to time = 1 subject |
| LSD/Psilocybin | A few times of reported use = 10 subjects |
| Opiates | A few times of reported use = 2 subjects |
| MDMA | A few times of reported use = 11 subjects |
| Genotype | Age (Mean ± SD) | p | NART IQ (Mean ± SD) | p | Number of Years of Education (Mean ± SD) | p |
|---|---|---|---|---|---|---|
| AKT1(G/G) (n = 19) | 26.5 ± 5.8 | NS | 98.8 ± 6.9 | NS | 17.5 ± 3.2 | NS |
| AKT1(G/A) (n = 9) | 26.3 ± 4.7 | 97.9 ± 5.3 | 16.6 ± 3.3 | |||
| AKT1(A/A) (n = 7) | 23.8 ± 6.6 | 95.5 ± 8.0 | 17.0 ± 7.4 |
| Cerebral Region | x | y | z | Size (No. of Voxels) | Side | p-Value |
|---|---|---|---|---|---|---|
| Inferior Frontal Gyrus | −43 | 26 | 4 | 14 | L | 0.006583 |
| −40 | 26 | −2 | 22 | L | ||
| −32 | 22 | −13 | 15 | L | ||
| −36 | 19 | −7 | 17 | L | ||
| Anterior Cingulate Gyrus | −4 | 22 | 37 | 10 | L | 0.002992 |
| −11 | 30 | 20 | 15 | L | ||
| Fusiform Gyrus | −22 | −67 | −7 | 8 | L | 0.002992 |
| Cuneus | −11 | −74 | 15 | 10 | L | 0.002992 |
| Middle Occipital Gyrus | −29 | −63 | 4 | 5 | L | 0.002992 |
| −25 | −81 | 9 | 25 | L | ||
| −18 | −85 | 15 | 8 | L | ||
| Culmen | −18 | −44 | −7 | 5 | L | 0.001496 |
| −22 | −44 | −18 | 7 | L | 0.008378 | |
| Middle Temporal Gyrus | −51 | −33 | −13 | 63 | L | 0.001496 |
| −51 | −11 | −13 | 7 | L | ||
| Brainstem, Midbrain | 0 | −15 | −18 | 5 | R | 0.001496 |
| Parahippocampal gyrus | −40 | −30 | −7 | 30 | L | 0.001496 |
| −25 | −26 | −18 | 12 | L | ||
| Cingulate Gyrus | −7 | 19 | 42 | 5 | L | 0.002992 |
| −11 | 26 | 26 | 14 | L | ||
| −11 | 26 | 31 | 10 | L | ||
| Anterior Cingulate | 0 | 30 | 15 | 20 | R | 0.002992 |
| −7 | 33 | 9 | 13 | L | ||
| 0 | 37 | 4 | 7 | R |
| Cerebral Region | x | y | z | Size (No. of Voxels) | Side | p-Value |
|---|---|---|---|---|---|---|
| Superior Frontal Gyrus | 36 | 52 | 15 | 27 | R | 0.003249 |
| Medial Frontal Gyrus | 40 | 48 | 9 | 11 | R | 0.003249 |
| 43 | 37 | 20 | 9 | R | ||
| 43 | 7 | 42 | 20 | R | 0.002067 | |
| 40 | 11 | 37 | 26 | R | ||
| 40 | 7 | 48 | 15 | R | ||
| 0 | −4 | 53 | 9 | R | 0.005907 | |
| Inferior Frontal Gyrus | 51 | 26 | 20 | 12 | R | 0.003249 |
| 43 | 4 | 31 | 17 | R | 0.002067 | |
| Sub-Gyral | 40 | 15 | 15 | 12 | R | 0.003249 |
| 11 | 30 | −2 | 8 | R | 0.002658 | |
| −43 | −33 | −7 | 18 | L | ||
| −47 | −15 | −13 | 5 | L | ||
| −25 | 7 | 42 | 26 | L | 0.005907 | |
| −29 | 7 | 37 | 10 | L | ||
| −32 | −4 | 37 | 6 | L | ||
| 25 | −4 | 53 | 23 | R | 0.007383 | |
| −29 | −70 | 26 | 5 | L | 0.009155 | |
| Middle Occipital Gyrus | −25 | −78 | 15 | 9 | L | 0.009155 |
| −25 | −74 | 9 | 8 | L | ||
| Middle Temporal Gyrus | −29 | −70 | 20 | 11 | L | 0.009155 |
| Precentral Gyrus | 29 | −15 | 48 | 21 | R | 0.007383 |
| Precuneus. | −36 | −70 | 37 | 5 | L | 0.009155 |
| Angular Gyrus | −36 | −74 | 31 | 6 | L | 0.009155 |
| Fusiform Gyrus. | −32 | −37 | −18 | 18 | L | 0.002658 |
| Midbrain | −4 | −26 | −13 | 47 | L | 0.002658 |
| Left Brainstem, Midbrain | −4 | −22 | −18 | 6 | L | 0.002658 |
| Insula | 40 | 15 | 9 | 13 | R | 0.003249 |
| Extra-Nuclear Corpus Callosum | 4 | 15 | 20 | 12 | R | 0.002658 |
| −4 | 26 | −2 | 8 | L | ||
| Amygdala/Parahippocampal gyrus | 22 | −7 | −18 | 36 | R | 0.003249 |
| Parahippocampal Gyrus | 29 | −22 | −13 | 23 | R | 0.003249 |
| Anterior Cingulate | 0 | 37 | 4 | 9 | R | 0.002658 |
| 4 | 33 | 9 | 6 | R | ||
| 0 | 30 | 15 | 19 | R |
| Cerebral Region | x | y | z | Size (No. of Voxels) | Side | p-Value |
|---|---|---|---|---|---|---|
| Middle Frontal Gyrus | 40 | 52 | 4 | 3 | R | 0.005075 |
| 36 | 48 | 9 | 7 | R | ||
| 36 | 48 | 15 | 21 | R | ||
| −36 | 44 | 9 | 17 | L | ||
| −40 | 41 | 15 | 13 | L | ||
| 40 | 37 | 20 | 8 | R | ||
| Medial Frontal Gyrus | 0 | 33 | 42 | 9 | R | 0.005075 |
| 0 | 30 | 37 | 15 | R | ||
| Inferior Frontal Gyrus | 40 | 26 | 4 | 7 | R | 0.005075 |
| 40 | 26 | −7 | 5 | R | ||
| −47 | 19 | 4 | 13 | L | ||
| Middle Temporal Gyrus | −51 | −11 | −13 | 7 | L | 0.007761 |
| −51 | −33 | −13 | 63 | L | ||
| Sub-Gyral | −32 | 44 | 4 | 12 | L | 0.005075 |
| −40 | −30 | −7 | 28 | L | 0.007761 | |
| −36 | −37 | −2 | 4 | L | ||
| Middle Occipital Gyrus | −22 | −85 | 15 | 9 | L | 0.007761 |
| −29 | −63 | 4 | 5 | L | ||
| Culmen. | −18 | −44 | −7 | 4 | L | 0.007761 |
| Cuneus | −11 | −74 | 15 | 10 | L | 0.007761 |
| −18 | −85 | 20 | 3 | L | ||
| Extra-Nuclear | −32 | 22 | −2 | 25 | L | 0.005075 |
| 32 | 22 | −2 | 7 | R | ||
| −32 | 15 | −7 | 25 | L | ||
| Left Brainstem, Midbrain | 0 | −15 | −18 | 4 | R | 0.007761 |
| Parahippocampal Gyrus | −25 | −26 | −18 | 18 | L | 0.007761 |
| Posterior Cingulate | −22 | −67 | 9 | 27 | L | 0.007761 |
| Cingulate Gyrus | −7 | 19 | 42 | 7 | L | 0.005075 |
| −11 | 26 | 26 | 15 | L | ||
| −11 | 26 | 31 | 14 | L | ||
| Anterior Cingulate | −11 | 26 | 20 | 17 | L | 0.005075 |
| 0 | 30 | 15 | 22 | R | ||
| −4 | 33 | 9 | 12 | L | ||
| 0 | 37 | 4 | 7 | R |
| Tal(x) | Tal(y) | Tal(z) | Cerebral Region |
|---|---|---|---|
| −22 | −50 | 0 | Parahippocampal Gyrus/Lingual Gyrus |
| −33 | −45 | −10 | Fusiform |
| −11 | −27 | −10 | Midbrain −> parahippocampal gyrus |
| −34 | −26 | −7 | Hippocampus −> Insula |
| −48 | −1 | −7 | Superior Temporal gyrus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blest-Hopley, G.; Colizzi, M.; Prata, D.; Giampietro, V.; Brammer, M.; McGuire, P.; Bhattacharyya, S. Epigenetic Mediation of AKT1 rs1130233’s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing. Brain Sci. 2021, 11, 1240. https://doi.org/10.3390/brainsci11091240
Blest-Hopley G, Colizzi M, Prata D, Giampietro V, Brammer M, McGuire P, Bhattacharyya S. Epigenetic Mediation of AKT1 rs1130233’s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing. Brain Sciences. 2021; 11(9):1240. https://doi.org/10.3390/brainsci11091240
Chicago/Turabian StyleBlest-Hopley, Grace, Marco Colizzi, Diana Prata, Vincent Giampietro, Michael Brammer, Philip McGuire, and Sagnik Bhattacharyya. 2021. "Epigenetic Mediation of AKT1 rs1130233’s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing" Brain Sciences 11, no. 9: 1240. https://doi.org/10.3390/brainsci11091240
APA StyleBlest-Hopley, G., Colizzi, M., Prata, D., Giampietro, V., Brammer, M., McGuire, P., & Bhattacharyya, S. (2021). Epigenetic Mediation of AKT1 rs1130233’s Effect on Delta-9-Tetrahydrocannabinol-Induced Medial Temporal Function during Fear Processing. Brain Sciences, 11(9), 1240. https://doi.org/10.3390/brainsci11091240