
Multimodal Role of PACAP in Glioblastoma


 ,
,  ,
,  ,
, 

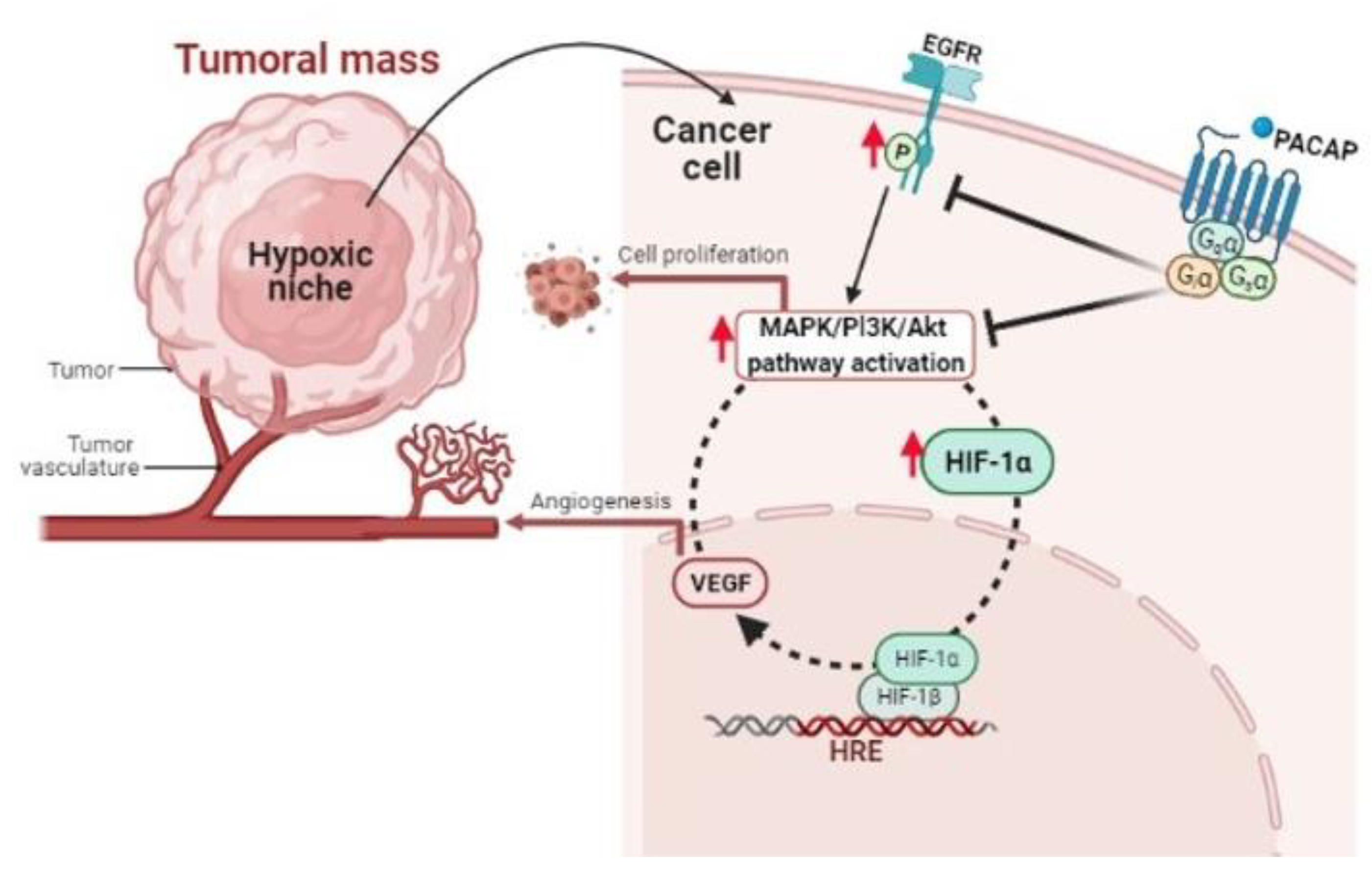{kind=link}
Abstract
1. Introduction
2. Glioblastoma Multiforme: Classification, Pathogenesis and Therapeutic Approaches
3. PACAP and Its Related Receptors in Cancer
4. PACAP Involvement in GBM Malignancy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Miyata, A.; Arimura, A.; Dahl, R.R.; Minamino, N.; Uehara, A.; Jiang, L.; Culler, M.D.; Coy, D.H. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 1989, 164, 567–574. [Google Scholar] [CrossRef]
- Cavallaro, S.; D’Agata, V.; Guardabasso, V.; Travali, S.; Stivala, F.; Canonico, P.L. Differentiation induces pituitary adenylate cyclase-activating polypeptide receptor expression in PC-12 cells. Mol. Pharmacol. 1995, 48, 56–62. [Google Scholar]
- D’Agata, V.; Cavallaro, S.; Stivala, F.; Canonico, P.L. Tissue-specific and developmental expression of pituitary adenylate cyclase-activating polypeptide (PACAP) receptors in rat brain. Eur. J. Neurosci. 1996, 8, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Canonico, P.L.; Copani, A.; D’Agata, V.; Musco, S.; Petralia, S.; Travali, S.; Stivala, F.; Cavallaro, S. Activation of pituitary adenylate cyclase-activating polypeptide receptors prevents apoptotic cell death in cultured cerebellar granule cells. Ann. N. Y. Acad. Sci. 1996, 805, 470–472. [Google Scholar] [CrossRef]
- Waschek, J.A. VIP and PACAP: Neuropeptide modulators of CNS inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef]
- Toth, D.; Szabo, E.; Tamas, A.; Juhasz, T.; Horvath, G.; Fabian, E.; Opper, B.; Szabo, D.; Maugeri, G.; D’Amico, A.G.; et al. Protective Effects of PACAP in Peripheral Organs. Front. Endocrinol. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Nuche-Berenguer, B.; Jensen, R.T. Vasoactive intestinal peptide/pituitary adenylate cyclase activating polypeptide, and their receptors and cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 38–47. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Reitano, R.; Magro, G.; Cavallaro, S.; Salomone, S.; D’Agata, V. PACAP and VIP Inhibit the Invasiveness of Glioblastoma Cells Exposed to Hypoxia through the Regulation of HIFs and EGFR Expression. Front. Pharmacol. 2016, 7, 139. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. PACAP and VIP regulate hypoxia-inducible factors in neuroblastoma cells exposed to hypoxia. Neuropeptides 2018, 69, 84–91. [Google Scholar] [CrossRef]
- Castorina, A.; Giunta, S.; Scuderi, S.; D’Agata, V. Involvement of PACAP/ADNP signaling in the resistance to cell death in malignant peripheral nerve sheath tumor (MPNST) cells. J. Mol. Neurosci. 2012, 48, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Scuderi, S.; D’Amico, A.G.; Drago, F.; D’Agata, V. PACAP and VIP increase the expression of myelin-related proteins in rat schwannoma cells: Involvement of PAC1/VPAC2 receptor-mediated activation of PI3K/Akt signaling pathways. Exp. Cell Res. 2014, 322, 108–121. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Scuderi, S.; Saccone, S.; Castorina, A.; Drago, F.; D’Agata, V. Antiproliferative effects of PACAP and VIP in serum-starved glioma cells. J. Mol. Neurosci. 2013, 51, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef]
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.U. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed. Pharmacother. 2017, 92, 681–689. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Teo, W.Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.G.; Li, X.; et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; De Maria, R.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in glioblastoma: Prognostic significance revisited. Neoplasia 2011, 13, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Bieńkowski, M.; Piaskowski, S.; Stoczyńska-Fidelus, E.; Szybka, M.; Banaszczyk, M.; Witusik-Perkowska, M.; Jesień-Lewandowicz, E.; Jaskólski, D.J.; Radomiak-Załuska, A.; Jesionek-Kupnicka, D.; et al. Screening for EGFR amplifications with a novel method and their significance for the outcome of glioblastoma patients. PLoS ONE 2013, 8, e65444. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Qi, S.; Lei, Y.; Si, G.; Ding, Y.; Han, H.; Zhang, X.; Wu, L.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar]
- Beiko, J.; Suki, D.; Hess, K.R.; Fox, B.D.; Cheung, V.; Cabral, M.; Shonka, N.; Gilbert, M.R.; Sawaya, R.; Prabhu, S.S.; et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-Oncol. 2014, 16, 81–91. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar] [CrossRef]
- Liang, B.C. Effects of hypoxia on drug resistance phenotype and genotype in human glioma cell lines. J. Neuro-Oncol. 1996, 29, 149–155. [Google Scholar] [CrossRef]
- Semenza, G.L. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell 2004, 5, 405–406. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro-Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene 2013, 32, 4057–4063. [Google Scholar] [CrossRef] [PubMed]
- Oka, N.; Soeda, A.; Inagaki, A.; Onodera, M.; Maruyama, H.; Hara, A.; Kunisada, T.; Mori, H.; Iwama, T. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2007, 360, 553–559. [Google Scholar] [CrossRef]
- Hamerlik, P.; Lathia, J.D.; Rasmussen, R.; Wu, Q.; Bartkova, J.; Lee, M.; Moudry, P.; Bartek, J., Jr.; Fischer, W.; Lukas, J.; et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 2012, 209, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef]
- Xu, H.; Rahimpour, S.; Nesvick, C.L.; Zhang, X.; Ma, J.; Zhang, M.; Zhang, G.; Wang, L.; Yang, C.; Hong, C.S.; et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: Implications for bevacizumab antiangiogenic therapy. Oncotarget 2015, 6, 11882–11893. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Weindel, K.; Moringlane, J.R.; Marmé, D.; Weich, H.A. Detection and quantification of vascular endothelial growth factor/vascular permeability factor in brain tumor tissue and cyst fluid: The key to angiogenesis? Neurosurgery 1994, 35, 439–448; discussion 448–449. [Google Scholar] [CrossRef]
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology 2015, 29, 720–727. [Google Scholar] [PubMed]
- Robles Irizarry, L.; Hambardzumyan, D.; Nakano, I.; Gladson, C.L.; Ahluwalia, M.S. Therapeutic targeting of VEGF in the treatment of glioblastoma. Expert Opin. Ther. Targets 2012, 16, 973–984. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncology 2009, 14, 1131–1138. [Google Scholar]
- Conroy, S.; Wagemakers, M.; Walenkamp, A.M.; Kruyt, F.A.; den Dunnen, W.F. Novel insights into vascularization patterns and angiogenic factors in glioblastoma subclasses. J. Neuro-Oncol. 2017, 131, 11–20. [Google Scholar] [CrossRef]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.; Henriksson, R.; et al. Patients with Proneural Glioblastoma May Derive Overall Survival Benefit From the Addition of Bevacizumab to First-Line Radiotherapy and Temozolomide: Retrospective Analysis of the AVAglio Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Bumes, E.; Rzonsa, S.; Hutterer, M.; Proescholdt, M.; Bogdahn, U.; Riemenschneider, M.J.; Uhl, M.; Wendl, C.; Hau, P. Adverse event grading following CTCAE v3.0 underestimates hypertensive side effects in patients with glioma treated with Bevacizumab. J. Neuro-Oncol. 2016, 127, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Hata, N.; Kuga, D.; Hatae, R.; Sangatsuda, Y.; Fujioka, Y.; Takigawa, K.; Mizoguchi, M. Update on Chemotherapeutic Approaches and Management of Bevacizumab Usage for Glioblastoma. Pharmaceuticals 2020, 13, 470. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Henry, V.; Sulman, E.; de Groot, J.F. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4392–4403. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C.; Cui, M.; Niu, J.; Ding, W. Inhibition of Bevacizumab-induced Epithelial-Mesenchymal Transition by BATF2 Overexpression Involves the Suppression of Wnt/β-Catenin Signaling in Glioblastoma Cells. Anticancer Res. 2017, 37, 4285–4294. [Google Scholar]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Sun, K.; Wang, X.; Pan, H.; Zhu, J.; Ji, X.; Li, X. Chrysin suppresses proliferation, migration, and invasion in glioblastoma cell lines via mediating the ERK/Nrf2 signaling pathway. Drug Des. Dev. Ther. 2018, 12, 721–733. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef]
- Agarwal, S.; Manchanda, P.; Vogelbaum, M.A.; Ohlfest, J.R.; Elmquist, W.F. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: Findings in an orthotopic rat xenograft model of glioma. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 33–39. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Kinzig, M.; Divé, I.; Maurer, G.D.; Filipski, K.; Harter, P.N.; Senft, C.; Bähr, O.; Hattingen, E.; Steinbach, J.P.; et al. Regorafenib CSF Penetration, Efficacy, and MRI Patterns in Recurrent Malignant Glioma Patients. J. Clin. Med. 2019, 8, 2031. [Google Scholar] [CrossRef] [PubMed]
- Indraccolo, S.; De Salvo, G.L.; Verza, M.; Caccese, M.; Esposito, G.; Piga, I.; Del Bianco, P.; Pizzi, M.; Gardiman, M.P.; Eoli, M.; et al. Phosphorylated Acetyl-CoA Carboxylase Is Associated with Clinical Benefit with Regorafenib in Relapsed Glioblastoma: REGOMA Trial Biomarker Analysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4478–4484. [Google Scholar] [CrossRef]
- Santangelo, A.; Rossato, M.; Lombardi, G.; Benfatto, S.; Lavezzari, D.; De Salvo, G.L.; Indraccolo, S.; Dechecchi, M.C.; Prandini, P.; Gambari, R.; et al. A molecular signature associated with prolonged survival in glioblastoma patients treated with regorafenib. Neuro-Oncol. 2021, 23, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Detti, B.; Scoccianti, S.; Lucidi, S.; Maragna, V.; Teriaca, M.A.; Ganovelli, M.; Desideri, I.; Lorenzetti, V.; Scoccimarro, E.; Greto, D.; et al. Regorafenib in glioblastoma recurrence: A case report. Cancer Treat. Res. Commun. 2021, 26, 100263. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Arimura, A.; Gozes, I.; Journot, L.; Laburthe, M.; Pisegna, J.R.; Rawlings, S.R.; Robberecht, P.; Said, S.I.; Sreedharan, S.P.; et al. International Union of Pharmacology. XVIII. Nomenclature of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Pharmacol. Rev. 1998, 50, 265–270. [Google Scholar]
- Arimura, A.; Somogyvári-Vigh, A.; Miyata, A.; Mizuno, K.; Coy, D.H.; Kitada, C. Tissue distribution of PACAP as determined by RIA: Highly abundant in the rat brain and testes. Endocrinology 1991, 129, 2787–2789. [Google Scholar] [CrossRef]
- Arimura, A.; Somogyvari-Vigh, A.; Weill, C.; Fiore, R.C.; Tatsuno, I.; Bay, V.; Brenneman, D.E. PACAP functions as a neurotrophic factor. Ann. N. Y. Acad. Sci. 1994, 739, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Arimura, A. Perspectives on pituitary adenylate cyclase activating polypeptide (PACAP) in the neuroendocrine, endocrine, and nervous systems. Jpn. J. Physiol. 1998, 48, 301–331. [Google Scholar] [CrossRef] [PubMed]
- Rivnyak, A.; Kiss, P.; Tamas, A.; Balogh, D.; Reglodi, D. Review on PACAP-Induced Transcriptomic and Proteomic Changes in Neuronal Development and Repair. Int. J. Mol. Sci. 2018, 19, 1020. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.N.; May, V.; Braas, K.M.; Hammack, S.E. PACAP orchestration of stress-related responses in neural circuits. Peptides 2021, 142, 170554. [Google Scholar] [CrossRef]
- Miyata, A.; Jiang, L.; Dahl, R.D.; Kitada, C.; Kubo, K.; Fujino, M.; Minamino, N.; Arimura, A. Isolation of a neuropeptide corresponding to the N-terminal 27 residues of the pituitary adenylate cyclase activating polypeptide with 38 residues (PACAP38). Biochem. Biophys. Res. Commun. 1990, 170, 643–648. [Google Scholar] [CrossRef]
- Segre, G.V.; Goldring, S.R. Receptors for secretin, calcitonin, parathyroid hormone (PTH)/PTH-related peptide, vasoactive intestinal peptide, glucagonlike peptide 1, growth hormone-releasing hormone, and glucagon belong to a newly discovered G-protein-linked receptor family. Trends Endocrinol. Metab. 1993, 4, 309–314. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Fahrenkrug, J.; Gozes, I.; Laburthe, M.; May, V.; Pisegna, J.R.; Vaudry, D.; Vaudry, H.; Waschek, J.A.; Said, S.I. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br. J. Pharmacol. 2012, 166, 4–17. [Google Scholar] [CrossRef]
- Blechman, J.; Levkowitz, G. Alternative Splicing of the Pituitary Adenylate Cyclase-Activating Polypeptide Receptor PAC1: Mechanisms of Fine Tuning of Brain Activity. Front. Endocrinol. 2013, 4, 55. [Google Scholar] [CrossRef]
- Jolivel, V.; Basille, M.; Aubert, N.; de Jouffrey, S.; Ancian, P.; Le Bigot, J.F.; Noack, P.; Massonneau, M.; Fournier, A.; Vaudry, H.; et al. Distribution and functional characterization of pituitary adenylate cyclase-activating polypeptide receptors in the brain of non-human primates. Neuroscience 2009, 160, 434–451. [Google Scholar] [CrossRef]
- Reglodi, D.; Kiss, P.; Horvath, G.; Lubics, A.; Laszlo, E.; Tamas, A.; Racz, B.; Szakaly, P. Effects of pituitary adenylate cyclase activating polypeptide in the urinary system, with special emphasis on its protective effects in the kidney. Neuropeptides 2012, 46, 61–70. [Google Scholar] [CrossRef]
- Reglodi, D.; Illes, A.; Opper, B.; Schafer, E.; Tamas, A.; Horvath, G. Presence and Effects of Pituitary Adenylate Cyclase Activating Polypeptide Under Physiological and Pathological Conditions in the Stomach. Front. Endocrinol. 2018, 9, 90. [Google Scholar] [CrossRef]
- Horvath, G.; Opper, B.; Reglodi, D. The Neuropeptide Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) is Protective in Inflammation and Oxidative Stress-Induced Damage in the Kidney. Int. J. Mol. Sci. 2019, 20, 4944. [Google Scholar] [CrossRef] [PubMed]
- Szegeczki, V.; Bauer, B.; Jüngling, A.; Fülöp, B.D.; Vágó, J.; Perényi, H.; Tarantini, S.; Tamás, A.; Zákány, R.; Reglődi, D.; et al. Age-related alterations of articular cartilage in pituitary adenylate cyclase-activating polypeptide (PACAP) gene-deficient mice. GeroScience 2019, 41, 775–793. [Google Scholar] [CrossRef]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Yon, L.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide and its receptors: From structure to functions. Pharmacol. Rev. 2000, 52, 269–324. [Google Scholar] [PubMed]
- Pisegna, J.R.; Wank, S.A. Cloning and characterization of the signal transduction of four splice variants of the human pituitary adenylate cyclase activating polypeptide receptor. Evidence for dual coupling to adenylate cyclase and phospholipase C. J. Biol. Chem. 1996, 271, 17267–17274. [Google Scholar] [CrossRef]
- Pantaloni, C.; Brabet, P.; Bilanges, B.; Dumuis, A.; Houssami, S.; Spengler, D.; Bockaert, J.; Journot, L. Alternative splicing in the N-terminal extracellular domain of the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor modulates receptor selectivity and relative potencies of PACAP-27 and PACAP-38 in phospholipase C activation. J. Biol. Chem. 1996, 271, 22146–22151. [Google Scholar] [CrossRef] [PubMed]
- Ushiyama, M.; Ikeda, R.; Sugawara, H.; Yoshida, M.; Mori, K.; Kangawa, K.; Inoue, K.; Yamada, K.; Miyata, A. Differential intracellular signaling through PAC1 isoforms as a result of alternative splicing in the first extracellular domain and the third intracellular loop. Mol. Pharmacol. 2007, 72, 103–111. [Google Scholar] [CrossRef]
- Pisegna, J.R.; Moody, T.W.; Wank, S.A. Differential signaling and immediate-early gene activation by four splice variants of the human pituitary adenylate cyclase-activating polypeptide receptor (hPACAP-R). Ann. N. Y. Acad. Sci. 1996, 805, 54–64; discussion 64–66. [Google Scholar] [CrossRef]
- Spengler, D.; Waeber, C.; Pantaloni, C.; Holsboer, F.; Bockaert, J.; Seeburg, P.H.; Journot, L. Differential signal transduction by five splice variants of the PACAP receptor. Nature 1993, 365, 170–175. [Google Scholar]
- Braas, K.M.; May, V. Pituitary adenylate cyclase-activating polypeptides directly stimulate sympathetic neuron neuropeptide Y release through PAC(1) receptor isoform activation of specific intracellular signaling pathways. J. Biol. Chem. 1999, 274, 27702–27710. [Google Scholar] [CrossRef][Green Version]
- Sokołowska, P.; Dejda, A.; Nowak, J.Z. Neuroprotective role of PACAP, VIP, and PHI in the central nervous system. Postepy Hig. Med. Dosw. (Online) 2004, 58, 416–427. [Google Scholar] [PubMed]
- Reglodi, D.; Lubics, A.; Tamás, A.; Szalontay, L.; Lengvári, I. Pituitary adenylate cyclase activating polypeptide protects dopaminergic neurons and improves behavioral deficits in a rat model of Parkinson’s disease. Behav. Brain Res. 2004, 151, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Jungling, A.; Longuespée, R.; Kriegsmann, J.; Casadonte, R.; Kriegsmann, M.; Juhasz, T.; Bardosi, S.; Tamas, A.; Fulop, B.D.; et al. Accelerated pre-senile systemic amyloidosis in PACAP knockout mice—A protective role of PACAP in age-related degenerative processes. J. Pathol. 2018, 245, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, G.; Iemmolo, R.; D’Amico, A.G.; La Cognata, V.; Costanzo, E.; Zappia, M.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. PACAP and PAC1R are differentially expressed in motor cortex of amyotrophic lateral sclerosis patients and support survival of iPSC-derived motor neurons. J. Cell. Physiol. 2018, 233, 3343–3351. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Morello, G.; Reglodi, D.; Cavallaro, S.; D’Agata, V. Differential Vulnerability of Oculomotor Versus Hypoglossal Nucleus During ALS: Involvement of PACAP. Front. Neurosci. 2020, 14, 805. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Musumeci, G.; Reglodi, D.; D’Agata, V. Effects of Pacap on Schwann Cells: Focus on Nerve Injury. Int. J. Mol. Sci. 2020, 21, 8233. [Google Scholar] [CrossRef]
- Solés-Tarrés, I.; Cabezas-Llobet, N.; Vaudry, D.; Xifró, X. Protective Effects of Pituitary Adenylate Cyclase-Activating Polypeptide and Vasoactive Intestinal Peptide Against Cognitive Decline in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 221. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Musumeci, G.; Reglodi, D.; D’Agata, V. PACAP and NAP: Effect of Two Functionally Related Peptides in Diabetic Retinopathy. J. Mol. Neurosci. 2021. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Saccone, S.; Federico, C.; Cavallaro, S.; Reglodi, D.; D’Agata, V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2943. [Google Scholar] [CrossRef]
- Yu, R.; Li, J.; Lin, Z.; Ouyang, Z.; Huang, X.; Reglodi, D.; Vaudry, D. TAT-tagging of VIP exerts positive allosteric modulation of the PAC1 receptor and enhances VIP neuroprotective effect in the MPTP mouse model of Parkinson’s disease. Biochim. Biophys. Acta. Gen. Subj. 2020, 1864, 129626. [Google Scholar] [CrossRef]
- Jungling, A.; Reglodi, D.; Maasz, G.; Zrinyi, Z.; Schmidt, J.; Rivnyak, A.; Horvath, G.; Pirger, Z.; Tamas, A. Alterations of Nigral Dopamine Levels in Parkinson’s Disease after Environmental Enrichment and PACAP Treatment in Aging Rats. Life 2021, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Jensen, R.T. Pituitary adenylate cyclase-activating polypeptide/vasoactive intestinal peptide [Part 1]: Biology, pharmacology, and new insights into their cellular basis of action/signaling which are providing new therapeutic targets. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 198–205. [Google Scholar] [CrossRef]
- Moody, T.W.; Jensen, R.T. Pituitary adenylate cyclase-activating polypeptide/vasoactive intestinal peptide (Part 2): Biology and clinical importance in central nervous system and inflammatory disorders. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Federico, C.; Saccone, S.; Morello, G.; La Cognata, V.; Cavallaro, S.; D’Agata, V. Molecular mechanisms involved in the protective effect of pituitary adenylate cyclase-activating polypeptide in an in vitro model of amyotrophic lateral sclerosis. J. Cell. Physiol. 2019, 234, 5203–5214. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J.A.; Dicicco-Bloom, E.M.; Lelievre, V.; Zhou, X.; Hu, Z. PACAP action in nervous system development, regeneration, and neuroblastoma cell proliferation. Ann. N. Y. Acad. Sci. 2000, 921, 129–136. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakamachi, T.; Endo, K.; Ito, K.; Machida, T.; Oka, T.; Hori, M.; Ishizaka, K.; Shioda, S. Distribution of pituitary adenylate cyclase-activating polypeptide (PACAP) in the human testis and in testicular germ cell tumors. Andrologia 2014, 46, 465–471. [Google Scholar] [CrossRef]
- Ferencz, S.; Reglodi, D.; Kaszas, B.; Bardosi, A.; Toth, D.; Vekony, Z.; Vicena, V.; Karadi, O.; Kelemen, D. PACAP and PAC1 receptor expression in pancreatic ductal carcinoma. Oncol. Lett. 2019, 18, 5725–5730. [Google Scholar] [CrossRef]
- Farini, D.; Puglianiello, A.; Mammi, C.; Siracusa, G.; Moretti, C. Dual effect of pituitary adenylate cyclase activating polypeptide on prostate tumor LNCaP cells: Short- and long-term exposure affect proliferation and neuroendocrine differentiation. Endocrinology 2003, 144, 1631–1643. [Google Scholar] [CrossRef]
- Lelièvre, V.; Pineau, N.; Du, J.; Wen, C.H.; Nguyen, T.; Janet, T.; Muller, J.M.; Waschek, J.A. Differential effects of peptide histidine isoleucine (PHI) and related peptides on stimulation and suppression of neuroblastoma cell proliferation. A novel VIP-independent action of PHI via MAP kinase. J. Biol. Chem. 1998, 273, 19685–19690. [Google Scholar] [CrossRef]
- Moody, T.W.; Chan, D.; Fahrenkrug, J.; Jensen, R.T. Neuropeptides as autocrine growth factors in cancer cells. Curr. Pharm. Des. 2003, 9, 495–509. [Google Scholar] [CrossRef]
- García-Fernández, M.O.; Bodega, G.; Ruíz-Villaespesa, A.; Cortés, J.; Prieto, J.C.; Carmena, M.J. PACAP expression and distribution in human breast cancer and healthy tissue. Cancer Lett. 2004, 205, 189–195. [Google Scholar] [CrossRef]
- García-Fernández, M.O.; Bodega, G.; Solano, R.M.; Ruíz-Villaespesa, A.; Sánchez-Chapado, M.; Carmena, M.J.; Prieto, J.C. Expression and distribution of pituitary adenylate cyclase-activating peptide in human prostate and prostate cancer tissues. Regul. Pept. 2002, 110, 9–15. [Google Scholar] [CrossRef]
- García-Fernández, M.O.; Collado, B.; Bodega, G.; Cortés, J.; Ruíz-Villaespesa, A.; Carmena, M.J.; Prieto, J.C. Pituitary adenylate cyclase-activating peptide/vasoactive intestinal peptide receptors in human normal mammary gland and breast cancer tissue. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2005, 20, 327–333. [Google Scholar] [CrossRef]
- Szanto, Z.; Sarszegi, Z.; Reglodi, D.; Nemeth, J.; Szabadfi, K.; Kiss, P.; Varga, A.; Banki, E.; Csanaky, K.; Gaszner, B.; et al. PACAP immunoreactivity in human malignant tumor samples and cardiac diseases. J. Mol. Neurosci. 2012, 48, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Tamas, A.; Javorhazy, A.; Reglodi, D.; Sarlos, D.P.; Banyai, D.; Semjen, D.; Nemeth, J.; Lelesz, B.; Fulop, D.B.; Szanto, Z. Examination of PACAP-Like Immunoreactivity in Urogenital Tumor Samples. J. Mol. Neurosci. 2016, 59, 177–183. [Google Scholar] [CrossRef]
- Bardosi, S.; Bardosi, A.; Nagy, Z.; Reglodi, D. Expression of PACAP and PAC1 Receptor in Normal Human Thyroid Gland and in Thyroid Papillary Carcinoma. J. Mol. Neurosci. 2016, 60, 171–178. [Google Scholar] [CrossRef]
- Vertongen, P.; d’Haens, J.; Michotte, A.; Velkeniers, B.; van Rampelbergh, J.; Svoboda, M.; Robberecht, P. Expression of pituitary adenylate cyclase activating polypeptide and receptors in human brain tumors. Peptides 1995, 16, 713–719. [Google Scholar] [CrossRef]
- Jaworski, D.M. Expression of pituitary adenylate cyclase-activating polypeptide (PACAP) and the PACAP-selective receptor in cultured rat astrocytes, human brain tumors, and in response to acute intracranial injury. Cell Tissue Res. 2000, 300, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, P.; Woussen-Colle, M.C.; Vertongen, P.; De Neef, P.; Hou, X.; Salmon, I.; Brotchi, J. Expression of pituitary adenylate cyclase activating polypeptide (PACAP) receptors in human glial cell tumors. Peptides 1994, 15, 661–665. [Google Scholar] [CrossRef]
- Reubi, J.C.; Läderach, U.; Waser, B.; Gebbers, J.O.; Robberecht, P.; Laissue, J.A. Vasoactive intestinal peptide/pituitary adenylate cyclase-activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer Res. 2000, 60, 3105–3112. [Google Scholar]
- Sharma, A.; Walters, J.; Gozes, Y.; Fridkin, M.; Brenneman, D.; Gozes, I.; Moody, T.W. A vasoactive intestinal peptide antagonist inhibits the growth of glioblastoma cells. J. Mol. Neurosci. 2001, 17, 331–339. [Google Scholar] [CrossRef]
- Barbarin, A.; Séité, P.; Godet, J.; Bensalma, S.; Muller, J.M.; Chadéneau, C. Atypical nuclear localization of VIP receptors in glioma cell lines and patients. Biochem. Biophys. Res. Commun. 2014, 454, 524–530. [Google Scholar] [CrossRef]
- Vertongen, P.; Camby, I.; Darro, F.; Kiss, R.; Robberecht, P. VIP and pituitary adenylate cyclase activating polypeptide (PACAP) have an antiproliferative effect on the T98G human glioblastoma cell line through interaction with VIP2 receptor. Neuropeptides 1996, 30, 491–496. [Google Scholar] [CrossRef]
- Sokolowska, P.; Nowak, J.Z. Effects of PACAP and VIP on cAMP-generating system and proliferation of C6 glioma cells. J. Mol. Neurosci. 2008, 36, 286–291. [Google Scholar] [CrossRef]
- Dufes, C.; Alleaume, C.; Montoni, A.; Olivier, J.C.; Muller, J.M. Effects of the vasoactive intestinal peptide (VIP) and related peptides on glioblastoma cell growth in vitro. J. Mol. Neurosci. 2003, 21, 91–102. [Google Scholar] [CrossRef][Green Version]
- Cochaud, S.; Chevrier, L.; Meunier, A.C.; Brillet, T.; Chadéneau, C.; Muller, J.M. The vasoactive intestinal peptide-receptor system is involved in human glioblastoma cell migration. Neuropeptides 2010, 44, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Cochaud, S.; Meunier, A.C.; Monvoisin, A.; Bensalma, S.; Muller, J.M.; Chadéneau, C. Neuropeptides of the VIP family inhibit glioblastoma cell invasion. J. Neuro-Oncol. 2015, 122, 63–73. [Google Scholar] [CrossRef]
- Bensalma, S.; Turpault, S.; Balandre, A.C.; De Boisvilliers, M.; Gaillard, A.; Chadéneau, C.; Muller, J.M. PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP. Cancers 2019, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Kubiatowski, T.; Jang, T.; Lachyankar, M.B.; Salmonsen, R.; Nabi, R.R.; Quesenberry, P.J.; Litofsky, N.S.; Ross, A.H.; Recht, L.D. Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J. Neurosurg. 2001, 95, 480–488. [Google Scholar] [CrossRef]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human cancers converge at the HIF-2alpha oncogenic axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Osefo, N.; Nuche-Berenguer, B.; Ridnour, L.; Wink, D.; Jensen, R.T. Pituitary adenylate cyclase-activating polypeptide causes tyrosine phosphorylation of the epidermal growth factor receptor in lung cancer cells. J. Pharmacol. Exp. Ther. 2012, 341, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Lee, L.; Iordanskaia, T.; Ramos-Alvarez, I.; Moreno, P.; Boudreau, H.E.; Leto, T.L.; Jensen, R.T. PAC1 regulates receptor tyrosine kinase transactivation in a reactive oxygen species-dependent manner. Peptides 2019, 120, 170017. [Google Scholar] [CrossRef]
- Moody, T.W.; Lee, L.; Jensen, R.T. The G Protein-Coupled Receptor PAC1 Regulates Transactivation of the Receptor Tyrosine Kinase HER3. J. Mol. Neurosci. 2020. [Google Scholar] [CrossRef]
- Grazia, M.; D’Amico, A.G.; Salvatore, S.; Concetta, F.; Maria, R.D.; Rosario, C.; Giuseppe, B.; Salvatore, G.; Giuseppe, M.; Velia, D. Effect of PACAP on hypoxia-induced angiogenesis and epithelial-mesenchymal transition in glioblastoma. Biomedicines. (under review).
- Roomi, M.W.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Modulation of MMP-2 and MMP-9 secretion by cytokines, inducers and inhibitors in human glioblastoma T-98G cells. Oncol. Rep. 2017, 37, 1907–1913. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Kanemura, Y.; Kohno, S.; Ohue, S.; Ozaki, S.; Matsumoto, S.; Suehiro, S.; et al. CD44 expression in the tumor periphery predicts the responsiveness to bevacizumab in the treatment of recurrent glioblastoma. Cancer Med. 2021, 10, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; Ohue, S.; et al. Hypoxia-induced phenotypic transition from highly invasive to less invasive tumors in glioma stem-like cells: Significance of CD44 and osteopontin as therapeutic targets in glioblastoma. Transl. Oncol. 2021, 14, 101137. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, A.G.; Maugeri, G.; Vanella, L.; Pittalà, V.; Reglodi, D.; D’Agata, V. Multimodal Role of PACAP in Glioblastoma. Brain Sci. 2021, 11, 994. https://doi.org/10.3390/brainsci11080994
D’Amico AG, Maugeri G, Vanella L, Pittalà V, Reglodi D, D’Agata V. Multimodal Role of PACAP in Glioblastoma. Brain Sciences. 2021; 11(8):994. https://doi.org/10.3390/brainsci11080994
Chicago/Turabian StyleD’Amico, Agata Grazia, Grazia Maugeri, Luca Vanella, Valeria Pittalà, Dora Reglodi, and Velia D’Agata. 2021. "Multimodal Role of PACAP in Glioblastoma" Brain Sciences 11, no. 8: 994. https://doi.org/10.3390/brainsci11080994
APA StyleD’Amico, A. G., Maugeri, G., Vanella, L., Pittalà, V., Reglodi, D., & D’Agata, V. (2021). Multimodal Role of PACAP in Glioblastoma. Brain Sciences, 11(8), 994. https://doi.org/10.3390/brainsci11080994