
Identification of a New Mutation in RSK2, the Gene for Coffin–Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
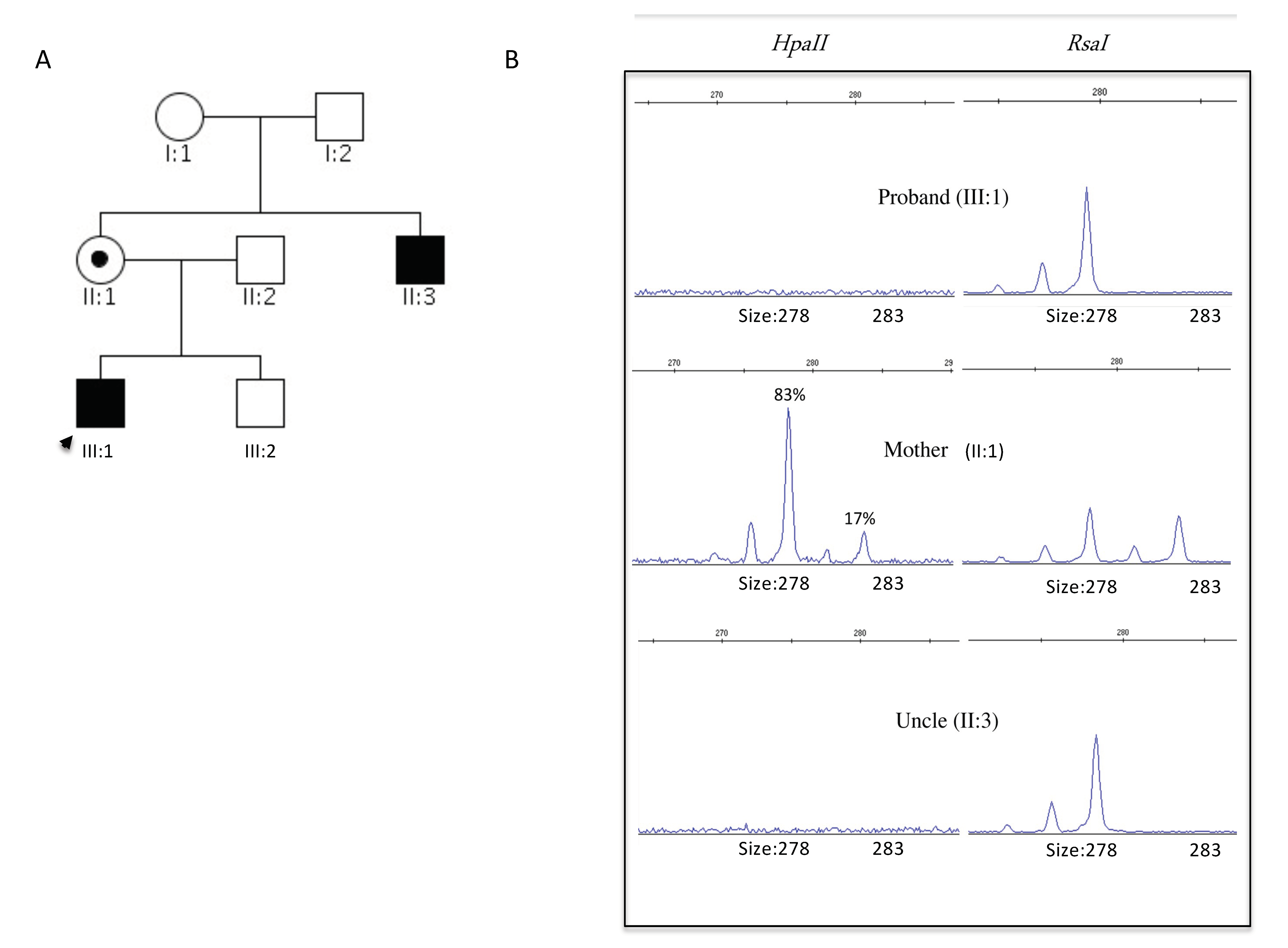
2.1. Clinics
2.2. Case 1 (III:1): The Proband
2.3. Case 2 (II:3): The Uncle
2.4. Genetics
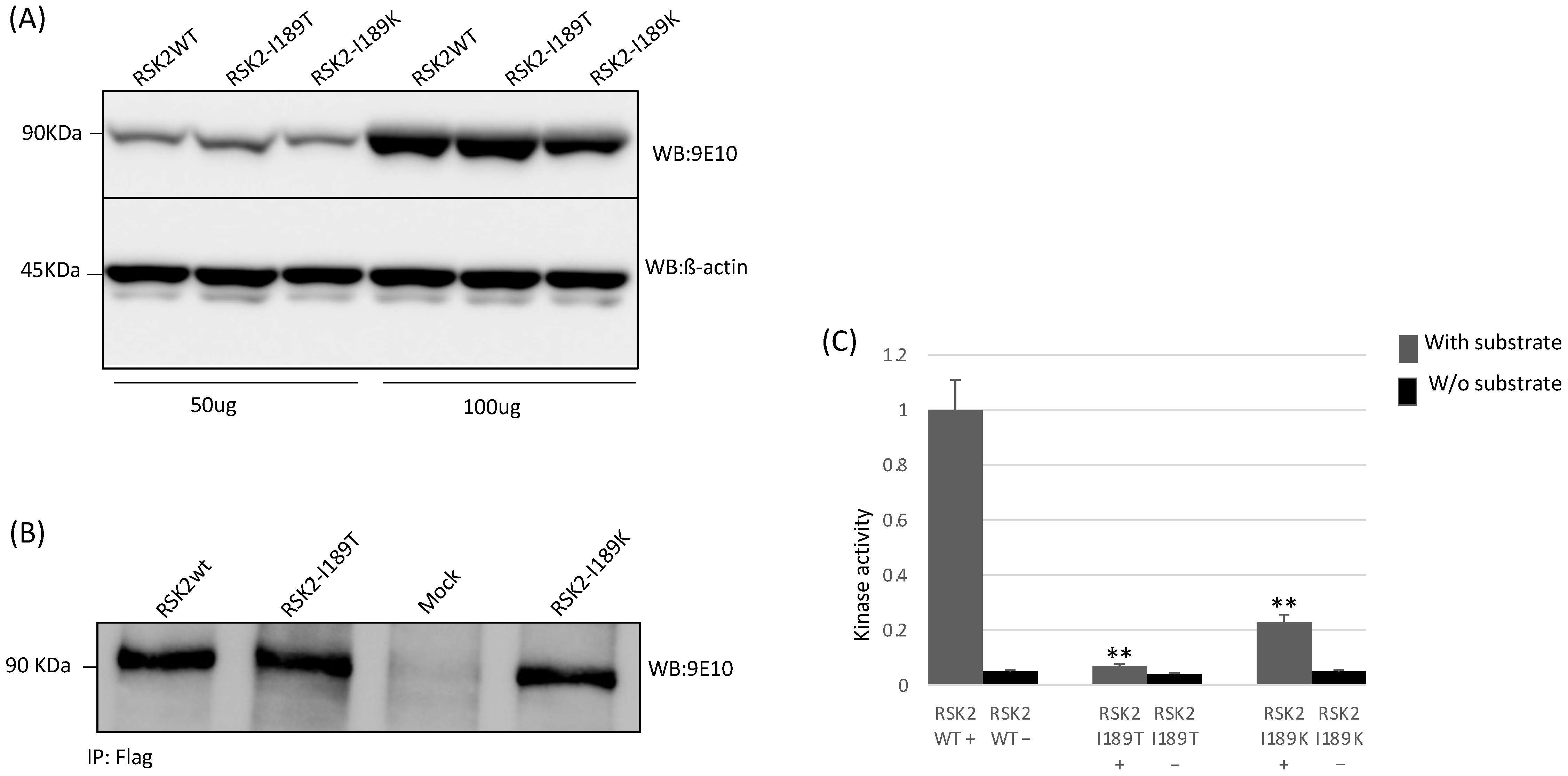
2.5. Functional Studies
3. Discussion
4. Conclusions
5. Material and Methods
5.1. Targeted Resequencing (TR)
5.2. X-Chromosome Inactivation (XCI)
5.3. DNA Cloning
5.4. Cell Culture and Transient Transfection
5.5. Western Blot Analysis
5.6. Immunoprecipitation and Protein Kinase Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CLS | Coffin–Lowry Syndrome |
| ID | Intellectual disability |
References
- Maystadt, I.; Destrée, A.; Benoit, V.; Aeby, A.; Lederer, D.; Moortgat, S.; Jurkiewicz, D.; Krajewska-Walasek, M.; Hanauer, A.; Thomas, G. RSK2mutation co-segregates with X-linked intellectual disability and attenuated Coffin-Lowry phenotype in a three-generation family. Clin. Genet. 2013, 85, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Lowry, B.; Miller, J.R.; Fraser, F.C. A New Dominant Gene Mental Retardation Syndrome. Am. J. Dis. Child. 1971, 121, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Manouvrier-Hanu, S.; Amiel, J.; Jacquot, S.; Merienne, K.; Moerman, A.; Coeslier, A.; Labarriere, F.; Vallee, L.; Croquette, M.F.; Hanauer, A. Unreported RSK2 missense mutation in two male sibs with an unusually mild form of Coffin-Lowry syndrome. J. Med. Genet. 1999, 36, 775–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temtamy, S.A.; Miller, J.D.; Hussels-Maumenee, I. The Coffin-Lowry syndrome: An inherited faciodigital mental retardation syndrome. J. Pediatr. 1975, 86, 724–731. [Google Scholar] [CrossRef]
- Hanauer, A.; Young, I.D. Coffin-Lowry syndrome: Clinical and molecular features. J. Med. Genet. 2002, 39, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, P.M.; Schneider, A.; Pannetier, S.; Heron, D.; Hanauer, A. Coffin–Lowry syndrome. Eur. J. Hum. Genet. 2009, 18, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, M.; Kornienko, M.; Byrne, N.; Reid, J.C.; Mizuarai, S.; Kotani, H.; Munshi, S.K. Crystal structures of the N-terminal kinase domain of human RSK1 bound to three different ligands: Implica-tions for the design of RSK1 specific inhibitors. Protein Sci. 2007, 16, 2626–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, G.; Lower, K.; White, S.; Delatycki, M.; Lampe, A.; Wright, M.; Smith, J.C.; Kerr, B.; Schelley, S.; Hoyme, H.; et al. The clinical picture of the Börjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin. Genet. 2004, 65, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Merienne, K.; Jacquot, S.; Pannetier, S.; Zeniou, M.; Bankier, A.; Gecz, J.; Mandel, J.L.; Mulley, J.C.; Sassone-Corsi, P.; Hanauer, A. A missense mutation in RPS6KA3 (RSK2) responsible for non-specific mental retardation. Nat. Genet. 1999, 22, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Tarpey, P.; Boyle, J.; Edkins, S.; Goodship, J.; Luo, Y.; Moon, J.; Teague, J.; Stratton, M.R.; A Futreal, P.; et al. Mutations in the RSK2(RPS6KA3) gene cause Coffin-Lowry syndrome and nonsyndromic X-linked mental retardation. Clin. Genet. 2006, 70, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, C.; Wang, Z.; Lv, H.; Li, X. In silico analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in the human GJA3 gene associated with congenital cataract. BMC Mol. Cell Biol. 2020, 21, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Morgutti, M.; Pecile, V.; Lenarduzzi, S.; Cappellani, S.; Falco, M.; Scarano, F.; Lonardo, F. A novel OTOA mutation in an Italian family with hearing loss. Gene Rep. 2017, 9, 111–114. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; De Pristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Lenarduzzi, S.; Morgan, A.; Faletra, F.; Cappellani, S.; Morgutti, M.; Mezzavilla, M.; Peruzzi, A.; Ghiselli, S.; Ambrosetti, U.; Graziano, C.; et al. Next generation sequencing study in a cohort of Italian patients with syndromic hearing loss. Hear. Res. 2019, 381, 107769. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Home|Research Square. Available online: https://www.researchsquare.com/ (accessed on 20 July 2021).
- Di Stazio, M.; Collesi, C.; Vozzi, D.; Liu, W.; Myers, M.; Morgan, A.; D’adamo, P.A.; Girotto, G.; Rubinato, E.; Giacca, M.; et al. TBL1Y: A new gene involved in syndromic hearing loss. Eur. J. Hum. Genet. 2019, 27, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stazio, M.; Morgana, A.; Brumat, M.; Bassani, S.; Dell’Orco, D.; Marino, V.; Garagnani, P.; Giuliani, C.; Girotto, P.G.G. New age-related hearing loss candidate genes in humans: An ongoing challenge. Gene 2020, 742, 144561. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stazio, M.; Bigoni, S.; Iuso, N.; Vuch, J.; Selvatici, R.; Ulivi, S.; d’Adamo, P.A. Identification of a New Mutation in RSK2, the Gene for Coffin–Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes. Brain Sci. 2021, 11, 1105. https://doi.org/10.3390/brainsci11081105
Di Stazio M, Bigoni S, Iuso N, Vuch J, Selvatici R, Ulivi S, d’Adamo PA. Identification of a New Mutation in RSK2, the Gene for Coffin–Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes. Brain Sciences. 2021; 11(8):1105. https://doi.org/10.3390/brainsci11081105
Chicago/Turabian StyleDi Stazio, Mariateresa, Stefania Bigoni, Nicola Iuso, Josef Vuch, Rita Selvatici, Sheila Ulivi, and Pio Adamo d’Adamo. 2021. "Identification of a New Mutation in RSK2, the Gene for Coffin–Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes" Brain Sciences 11, no. 8: 1105. https://doi.org/10.3390/brainsci11081105
APA StyleDi Stazio, M., Bigoni, S., Iuso, N., Vuch, J., Selvatici, R., Ulivi, S., & d’Adamo, P. A. (2021). Identification of a New Mutation in RSK2, the Gene for Coffin–Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes. Brain Sciences, 11(8), 1105. https://doi.org/10.3390/brainsci11081105