
Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Patients and DNA Samples’ Preparation
2.2. Exome Sequencing
3. Results
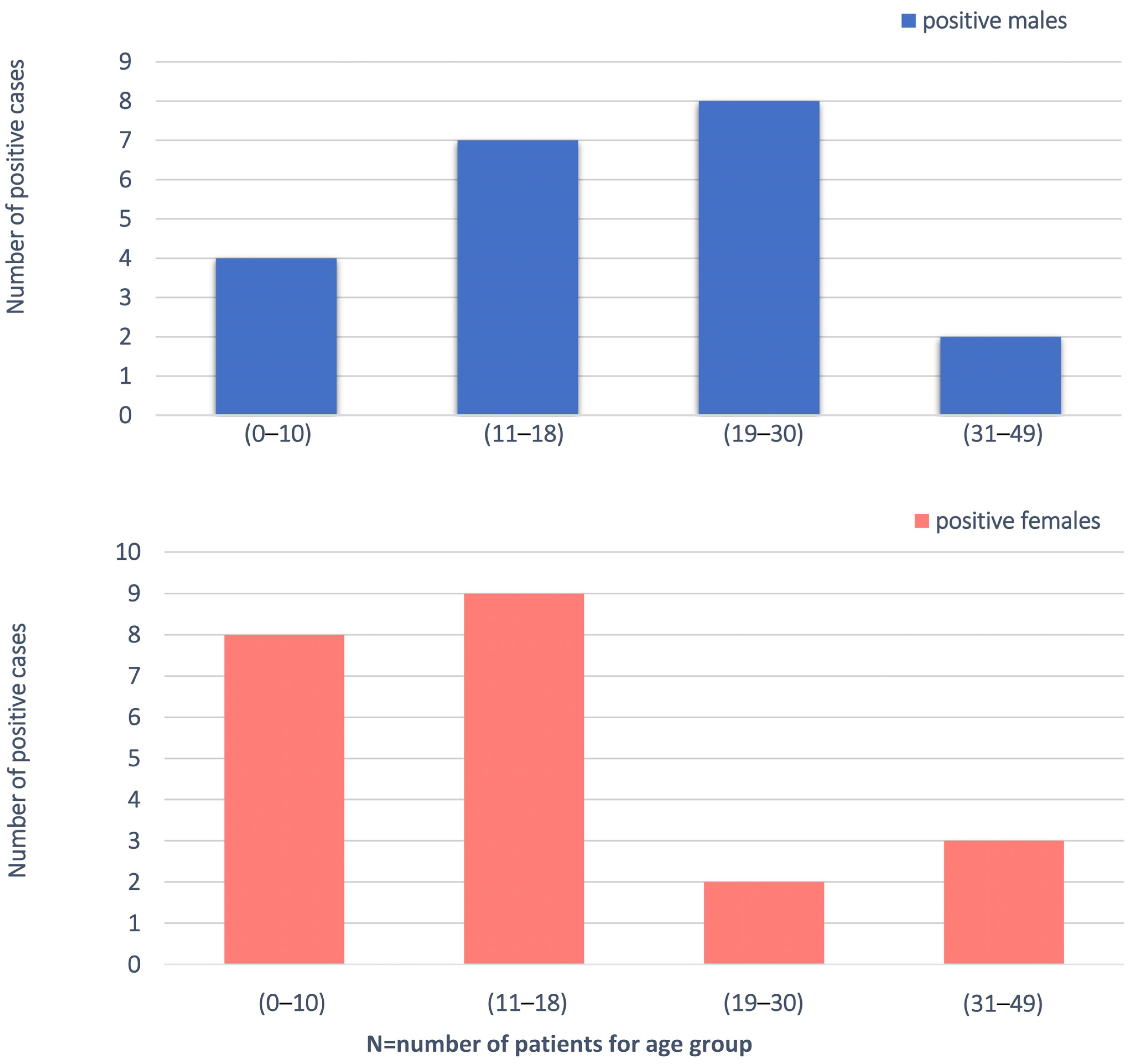
3.1. Clinical Characteristics of Patients
3.2. P/LP Variants Identified by ES
3.3. Uncertain Variants Identified by ES
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tassé, M.J.; Luckasson, R.; Nygren, M. AAIDD proposed recommendations for ICD-11 and the condition previously known as mental retardation. Intellect. Dev. Disabil. 2013, 51, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O. Intellectual disability and nutrition-related health. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef]
- Maulik, P.K.; Mascarenhas, M.N.; Mathers, C.D.; Dua, T.; Saxena, S. Prevalence of intellectual disability: A meta-analysis of population-based studies. Res. Dev. Disabil. 2011, 32, 419–436. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Kiani, A.K.; Miertus, J.; Barati, S.; Manara, E.; Paolacci, S.; Stuppia, L.; Gurrieri, F.; Bertelli, M. Genetic analysis of intellectual disability and autism. Acta Biomed. 2020, 91, e2020003. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.R.; Gonda, X.; Tarazi, F.I. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol. Ther. 2018, 190, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of autism spectrum disorder among children aged 8 Years—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Zablotsky, B.; Black, L.I.; Maenner, M.J.; Schieve, L.A.; Danielson, M.L.; Bitsko, R.H.; Blumberg, S.J.; Kogan, M.D.; Boyle, C.A. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics 2019, 144, e20190811. [Google Scholar] [CrossRef] [PubMed]
- Baranova, J.; Dragunas, G.; Botellho, M.C.S.; Ayub, A.L.P.; Bueno-Alves, R.; Alencar, R.R.; Papaiz, D.D.; Sogayar, M.C.; Ulrich, H.; Correa, R.G. Autism Spectrum Disorder: Signaling Pathways and Prospective Therapeutic Targets; Springer: Berlin/Heidelberg, Germany, 2021; Volume 41, ISBN 0123456789. [Google Scholar]
- Thevenon, J.; Duffourd, Y.; Masurel-Paulet, A.; Lefebvre, M.; Feillet, F.; El Chehadeh-Djebbar, S.; St-Onge, J.; Steinmetz, A.; Huet, F.; Chouchane, M.; et al. Diagnostic odyssey in severe neurodevelopmental disorders: Toward clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet. 2016, 89, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Mol. Autism 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.; Fennell, T.; Carneiro, M.; Van der Auwera, G.; Kling, D.; Gauthier, L.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.; Carlson, M. Whole exome sequencing in pediatric neurology patients: Clinical implications and estimated cost analysis. J. Child Neurol. 2016, 31, 887–894. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Doccini, V.; Bernardini, L.; Novelli, A.; Loddo, S.; Capalbo, A.; Filippi, T.; Carey, J.C. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur. J. Paediatr. Neurol. 2013, 17, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, F.R.; McPartland, J.C. From Kanner to DSM-5: Autism as an evolving diagnostic concept. Annu. Rev. Clin. Psychol. 2014, 10, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef]
- Renieri, A.; Meloni, I.; Longo, I.; Ariani, F.; Mari, F.; Pescucci, C.; Cambi, F. Rett syndrome: The complex nature of a monogenic disease. J. Mol. Med. 2003, 81, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U. Rett syndrome is caused by mutations in X-linked. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 2, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Snijders Blok, L.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.F.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on wnt signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef]
- Wang, X.; Posey, J.E.; Rosenfeld, J.A.; Bacino, C.A.; Scaglia, F.; Immken, L.D.; Harris, J.M.; Hickey, S.E.; Mosher, T.M.; Slavotinek, A.; et al. Phenotypic expansion in DDX3X—A common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 2018, 15, 1277–1285. [Google Scholar] [CrossRef]
- Gilling, M.; Rasmussen, H.B.; Calloe, K.; Sequeira, A.F.; Baretto, M.; Oliveira, G.; Almeida, J.; Lauritsen, M.B.; Ullmann, R.; Boonen, S.E.; et al. Dysfunction of the heteromeric KV7.3/KV7.5 potassium channel is associated with autism spectrum disorders. Front. Genet. 2013, 4, 54. [Google Scholar] [CrossRef]
- Nemani, T.; Steel, D.; Kaliakatsos, M.; DeVile, C.; Ververi, A.; Scott, R.; Getov, S.; Sudhakar, S.; Male, A.; Mankad, K.; et al. KIF1A-related disorders in children: A wide spectrum of central and peripheral nervous system involvement. J. Peripher. Nerv. Syst. 2020, 25, 117–124. [Google Scholar] [CrossRef]
- Garde, A.; Cornaton, J.; Sorlin, A.; Moutton, S.; Nicolas, C.; Juif, C.; Geneviève, D.; Perrin, L.; Khau-Van-Kien, P.; Smol, T.; et al. Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations. Clin. Genet. 2021. [Google Scholar] [CrossRef]
- Assia Batzir, N.; Posey, J.E.; Song, X.; Akdemir, Z.C.; Rosenfeld, J.A.; Brown, C.W.; Chen, E.; Holtrop, S.G.; Mizerik, E.; Nieto Moreno, M.; et al. Phenotypic expansion of POGZ-related intellectual disability syndrome (White-Sutton syndrome). Am. J. Med. Genet. Part A 2020, 182, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Precht, K.S.; Lese, C.M.; Spiro, R.P.; Huttenlocher, P.R.; Johnston, K.M.; Baker, J.C.; Christian, S.L.; Kittikamron, K.; Ledbetter, D.H. Two 22q telomere deletions serendipitously detected by FISH. J. Med. Genet. 1998, 35, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.; Prasad, A.N.; Chodirker, B.N.; Lee, C.; Dawson, A.K.; Jocelyn, L.J.; Chudley, A.E. Genetic evaluation of pervasive developmental disorders: The terminal 22q13 deletion syndrome may represent a recognizable phenotype. Clin. Genet. 2000, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Powers, P.A.; Scherer, S.W.; Tsui, L.C.; Gregg, R.G.; Hogan, K. Localization of the Gene Encoding the α2/δ Subunit (CACNL2A) of the Human Skeletal Muscle Voltage-Dependent Ca2+ Channel to Chromosome 7q21-q22 by Somatic Cell Hybrid Analysis. Genomics 1994, 19, 192–193. [Google Scholar] [CrossRef][Green Version]
- Vergult, S.; Dheedene, A.; Meurs, A.; Faes, F.; Isidor, B.; Janssens, S.; Gautier, A.; Le Caignec, C.; Menten, B. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur. J. Hum. Genet. 2015, 23, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Li, Z.; Offord, J.; Davis, M.D.; McCormick, J.; Taylor, C.P.; Walker, L.C. Calcium channel alpha2-delta type 1 subunit is the major binding protein for pregabalin in neocortex, hippocampus, amygdala, and spinal cord: An ex vivo autoradiographic study in alpha2-delta type 1 genetically modified mice. Brain Res. 2006, 1075, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Sugo, T.; Abe, M.; Shimomura, Y.; Kurihara, M.; Kitada, C.; Kikuchi, K.; Shintani, Y.; Kurokawa, T.; Onda, H.; et al. Urotensin II is the endogenous ligand of a G-protein-coupled orphan receptor, SENR (GPR14). Biochem. Biophys. Res. Commun. 1999, 137, 579–588. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Patient | Gene | Variant (HGVS) | Protein (HGVS) | Gender | Age (Years) | ID/ASD | Craniofacial Dysmorphisms | Additional Clinical Signs | OMIM Phenotype (n°) |
|---|---|---|---|---|---|---|---|---|---|
| #1 | ADNP | c.2314dup | p.(Thr772Asnfs *16) | Male | 7 | ID | Yes (Plagiocephaly, prominent forehead, sparse eyebrows, broad nasal bridge, small ears, uplifted earlobes, conical teeth) | Bilateral clinodactyly of the 5th toe, supernumerary nipple, epilepsy, heteroaggressive behaviour | Helsmoortel-van der Aa syndrome (#615873) |
| #2 | AP4M1 | c.1257_1282del c.1317G > A | p.(Val421Alafs *98) p.(Trp439 *) | Female | 18 | ID | Yes (microcephaly, triangular face, widow’s peak, thick eyebrows, large nose, flat philtrum, thin lips, high vaulted and narrow palate) | Short stature, clinodactyly of the fifth finger, laryngomalacia, splenium thinning, epilepsy, hearing loss, flat foot, stereotypies, epilepsy, speech defect | Spastic paraplegia 50, autosomal recessive (#612936) |
| #3 | ATP1A3 | c.2116G > A | p.(Gly706Arg) | Female | 22 | ID | No | Epilepsy, ataxia, limited speech | Alternating hemiplegia of childhood 2 (#614820)/CAPOS syndrome (#601338)/Dystonia-12 (#128235) |
| #4 | BCOR | c.776C > T | p.(Ser259Leu) | Male | 14 | ID and ASD | Yes (low hairline, periorbital swelling, big mouth, full lips) | Stereotypies | Microphthalmia, syndromic, 2 (#300166) |
| #5 | BSCL2 | c.856C > T | p.(Arg286 *) | Female | 22 | ID | Yes (elongated facies, prominent ears, high nasal bridge, open mouth and protruded tongue) | Dystonia, myoclonia, drug-resistant epilepsy, language delay, psychomotor delay, bruxism, hypertrichosis | Encephalopathy, progressive, with or without lipodystrophy (#615924) |
| #6 | BSCL2 | c.856C > T | p.(Arg286 *) | Female | 16 | ID | Yes (facies with coarse features, synophria, bulbous nose, large ears, large mouth) | Dystonia, myoclonia, hypertrichosis, language delay, drug-resistant epilepsy, psychomotor delay | Encephalopathy, progressive, with or without lipodystrophy (#615924) |
| #7 | CACNA1A | c.4055G > A | p.(Arg1352Gln) | Male | 14 | ID | Yes (Craniosynostosis, long filter, long eyelashes, thin upper lip) | Stereotypies, alternating hemiplegia, language delay | Developmental and epileptic encephalopathy 42 (#617106); Episodic ataxia, type 2 (#108500); Migraine, familial hemiplegic, 1 (#141500); Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia (#141500); Spinocerebellar ataxia 6 (#183086) |
| #8 | CSNK2B | c.170del | p.(Glu57Glyfs *15) | Female | 20 | ID | Yes (Open mouth, protruding tongue, narrow palate, dental anomalies) | Epilepsy, hypotonia, developmental delay, absent language | Poirier-Bienvenu neurodevelopmental syndrome (#618732) |
| #9 | CTU2 | c.881C > A | p.(Ser294 *) | Female | 22 | ID | Yes (broad eyebrows, low anterior and posterior hairline, forehead hypertrichosis, prominent ears, eye asymmetry, large nose with wide nostrils, large mouth, dental anomalies) | Upper limb hypotonia, lower limb hypertonia, recurrent infections, thinning corpus callosum, growth retardation, pectus excavatum, clinodactyly of the 5th fingers. | Microcephaly, facial dysmorphism, renal agenesis, and ambiguous genitalia syndrome (#617057) |
| #10 | DEPDC5 | c.1453C > T | p.(Arg485 *) | Male | 11 | ID | Yes (low hairline, flattened nasal bridge, bulbous nasal tip, anteverted nostrils, full lips, small and widely spaced teeth, anteverted ears) | Epilepsy, language delay | Epilepsy, familial focal, with variable foci 1 (#604364) |
| #11 | DDX3X | c.976C > T | p.(Arg326Cys) | Female | 12 | ID and ASD | Yes (Microcephaly, long face, smooth and long philtrum, strabismus, up slanting palpebral fissures) | Developmental delay, stereotypic hand movements, hypotonia, bruxism, sialorrhea, corpus callosum hypoplasia | Intellectual developmental disorder, X-linked, syndrome, Snijders Blok type (#300958) |
| #12 | DYRK1A | c.1669C > T | p.(Gln557 *) | Male | 16 | ID | Yes (Microcephaly, narrow forehead, frontal bossing, depressed nasal bridge, short philtrum, prognathism) | Hypotonia, epilepsy, stereotypies, absent language | Mental retardation, autosomal dominant 7 (#614104) |
| #13 | EFTUD2 | c.702 + 1G > A | NA | Female | 22 | ID | Yes (prominent columella, sloping forehead, high and scattered eyebrows, deeply set eyes, nose with anteverted nostrils, narrow palate, prominent incisors, prominent ears) | Hypertrichosis, small hands and feet, joint hyperextensivity | Mandibulofacial dysostosis, Guion-Almeida type (#603892) |
| #14 | FGFR3 | c.749C > G | p.(Pro250Arg) | Female | 4 | ID and ASD | Yes (craniosynostosis, flat forehead, down slanting palpebral fissures, high nasal bridge) | Language delay, hyperchromic and hypochromic spots | Muenke syndrome (#602849) CATSHL syndrome (#610474) |
| #15 | HK1 | c.1367C > T | p.(Thr456Met) | Male | 16 | ID | Yes (proptosis, long eyelashes, synophria, narrow palate, dental anomalies, small mouth, and full lips) | Bilateral cryptorchidism, psychomotor delay, poor vision, ventricular system dilation, immature hippocampal structures, spastic paraparesis, epilepsy, hypothonia, absent language | Neurodevelopmental disorder with visual defects and brain anomalies (#618547) |
| #16 | IQSEC2 | c.3780del | p.(Gln1261Serfs *136) | Female | 22 | ID | No | Strabismus, cerebral atrophy, sialorrhea, motor problems, bruxism | Mental retardation, X-linked 1/78 (#309530) |
| #17 | KANSL1 | c.985_986del | p.(Leu329Glufs *22) | Female | 6 | ID | Yes (cleft palate, deeply set eyes, high nasal bridge, bulbous nasal tip) | Language delay, epilepsy, aggressiveness | Koolen-De Vries syndrome (#610443) |
| #18 | KCNQ3 | c.688C > T | p.(Arg230Cys) | Male | 39 | ID and ASD | Yes (triangular facies, high nasal bridge, bulbous nasal tip, open mouth, micrognathia, narrow and downturned eyelids) | Stereotypies, aggressiveness, bladder anomalies | Seizures, benign neonatal, 2 (#121201) |
| #19 | KIF1A | c.37C > T | p.(Arg13Cys) | Female | 16 | ID | No | Spastic paraparesis, behaviour disorder, slight enlargement of the interfolial spaces of the cerebellar hemispheres, hypertone | Spastic paraplegia 30, autosomal dominant (#610357) |
| #20 | KIF1A | c.914C > T | p.(Pro305Leu) | Female | 10 | ID | Yes (sparse eyebrows, small nose, thin upper lip, dental anomalies, chubby cheeks) | Language delay, cerebellar and worm atrophy, psychomotor delay, brain abnormalities, hypertrichosis, bilateral clinodactyly of the second/third/fourth/fifth toes | Spastic paraplegia 30, autosomal dominant (#610357) |
| #21 | KIF1A | c.914C > T | p.(Pro305Leu) | Male | 49 | ID | No | Ataxia, spastic paraparesis, angioma, nystagmus, epilepsy | Spastic paraplegia 30, autosomal dominant (#610357) |
| #22 | KMT2A | c.5256delA | p.(Ala1753Profs*70) | Male | 16 | ID | Yes (thick eyebrows, narrow and short eyelids, depressed helix, nose with bulbous tip, thick lips) | Hypertrichosis of the legs, arms and lumbar region, large hands, short fifth ray of the foot | Wiedemann-Steiner syndrome (#605130) |
| #23 | MBOAT7 | c.477C > G | p.(Tyr159 *) | Female | 2 | ID | No | Motor stereotypies, epilepsy, thinning of the corpus callosum, ventricular enlargement | Mental retardation, autosomal recessive 57 (#617188) |
| #24 | MED13L | c.72 + 1G > T | NA | Female | 49 | ID | Yes (sparse eyebrows, hypertelorism, narrow eyelids, gingival hypertrophy) | Syndactyly of the second and third toes, small feet, hypertrophy of the limbs and truncal obesity, strabismus | Mental retardation and distinctive facial features with or without cardiac defects (#616789) |
| #25 | MMACHC | c.440G > C | p.(Gly147Ala) | Male | 12 | ID | Yes (synophria, horizontal eyebrows, wide nasal tip, anteverted nostrils, long filter, dental anomalies) | Spastic paraparesis, language delay, polyneuropathy | Methylmalonic aciduria and homocystinuria, cblC type (#277400) |
| #26 | POGZ | c.1180_1181del | p.(Met394Valfs *9) | Female | 6 | ID | Yes (microcephaly, deeply set eyes, nose with bulbous tip, anteverted nostrils, full lips) | Hyperactivity, blepharophimosis, brachydactyly, nail hypoplasia, kidney abnormalities, language delay | White-Sutton syndrome (#616364) |
| #27 | POGZ | c.1180_1181del | p.(Met394Valfs *9) | Male | 12 | ID | Yes (narrow bitemporal diameter, narrow and upward eyelid rims, deep philtrum, progatism, exaggerated Cupid′s bow, buccal rim pointing downwards, uplifted ear lobe) | Hypotonia, obesity | White-Sutton syndrome (#616364) |
| #28 | POGZ | c.1180_1181del | p.(Met394Valfs *9) | Female | 44 | ID | No | Microcephaly, brachydactyly, nail hypoplasia | White-Sutton syndrome (#616364) |
| #29 | PTPN11 | c.1471C > A | p.(Pro491Thr) | Male | 2 | ID | Yes (high forehead, low-set ears with large, downward-pointing auricles, down-slanting eyelids, broad nasal tip, long and thick filter, exaggerated Cupid′s bow) | Café au lait spots in the thoracic and lumbar region, nail hypoplasia of the fifth toe, neurodevelopmental delay, cryptorchidism | Noonan syndrome 1 (#163950) |
| #30 | RHOBTB2 | c.1382G > A | p.(Arg461His) | Male | 18 | ID | Yes (microcephaly, thick eyebrows, deeply set eyes, square chin) | epilepsy, thinning corpus callosum, language delay, cold extremities, cerebellar atrophy, muscle hypotrophy | Developmental and epileptic encephalopathy 64 (# 618004) |
| #31 | SHANK3 | c.2717_2718dup | p.(Ser907Alafs *3) | Male | 45 | ID and ASD | Yes (alopecia in the fronto-temporal region, depressed ocular region, thin upper lip) | Absent language, obsessive behaviour, tapered fingers with widening of the intermediate interphalangeal joints, nail dystrophy, clinodactyly of the third toe, stereotypies, mouth chewing automatisms | Phelan-McDermid syndrome (#606232) |
| #32 | SHANK3 | c.2313 + 1G > A | NA | Male | 21 | ID and ASD | No | epilepsy, psychomotor delay | Phelan-McDermid syndrome (#606232) |
| #33 | SHANK3 | c.3250_3253del | p.(Leu1084Cysfs *9) | Male | 18 | ID | No | Language delay, psychomotor delay, manual stereotypies, increased tolerance to pain | Phelan-McDermid syndrome (#606232) |
| #34 | SPG7 | c.233T > A | p.(Leu78 *) | Female | 10 | ID | Yes (high forehead, spaced teeth, pursed lips attitude) | Epilepsy, neurodevelopmental regression, stereotypies, syndactyly | Spastic paraplegia 7, autosomal recessive (#607259) |
| #35 | SPTBN2 | c.1310G > A | p.(Arg437Gln) | Male | 17 | ID | Yes (sloping forehead, prognathism) | Strabismus, hyperchromic spot in the thoracic area, cerebellar atrophy, language delay, ataxia | Spinocerebellar ataxia 5 (#600224) |
| #36 | SYNGAP1 | c.2337–1G > A | NA | female | 26 | ID and ASD | No | Stereotypies, self-harm, cold extremities, sphincter control not acquired, language regression | Mental retardation, autosomal dominant 5 (#612621) |
| #37 | TBCE | c.464T > A c.134dupA | p.(Ile155Asn) p.(Arg46Glufs *5) | Male | 21 | ID | Yes (bulbous nasal tip, full lips, spaced teeth) | Scoliosis, sialorrhea, optic atrophy, psychomotor delay, epilepsy, spastic paraparesis, absent language. | Encephalopathy, progressive, with amyotrophy and optic atrophy (#617207) |
| #38 | TREX1 | c.558_573del | p.(Phe186Leufs *24) | Male | 10 | ID and ASD | No | Gastroesophageal reflux, sphincter control not acquired, stereotyped behaviour | Aicardi-Goutieres syndrome 1 dominant and recessive (#225750) |
| #39 | TUBA1A | c.352G > A | p.(Val118Met) | Male | 9 | ID | Yes (advanced hairline, long and thick eyebrows, anteverted nostrils, long filter, thin upper lip) | Hypoplasia of the cerebellar vermis, thinned corpus callosum, cerebellar asymmetry, angioma, hypertrichosis, epilepsy, apraxia, ataxia, and psychomotor delay | Lissencephaly 3 (#611603) |
| #40 | UPF3B | c.1288C > T | p.(Arg430 *) | Male | 7 | ID and ASD | Yes (Wide forehead, arched eyebrows, deeply set eyes, pointed chin) | Limited speech, neurodevelopmental delay | Mental retardation, X-linked syndromic 14 (#300676) |
| #41 | WDR45 | c.66del | p.(Cys23Alafs *15) | Female | 20 | ID | Yes (thick eyebrows, prominent upper arch, hyperemic gums, high palate) | Limited speech, scoliosis, locomotor impairment, manual stereotypies, tapered fingers | Neurodegeneration with brain iron accumulation 5 (#300894) |
| #42 | WFS1 | c.124C > T | p.(Arg42 *) | Male | 6 | ID and ASD | No | Language delay, oppositional and provocative behaviour | Wolfram-like syndrome, autosomal dominant (#614296) |
| #43 | WFS1 | c.1230_1233del | p.(Leu412Serfs *29) | Female | 14 | ID | Yes (microcephaly, synophria, long eyelashes, bulbous nasal tip) | Hypertrichosis, drug-resistant seizures, spastic tetraparesis, renal failure. | Wolfram-like syndrome, autosomal dominant (#614296) |
| Patient | Gene | Variant (HGVS) | Protein (HGVS) | Gender | Age (Years) | ID/ASD | Craniofacial Dysmorphisms | Additional Clinical Signs |
|---|---|---|---|---|---|---|---|---|
| #44 | CACNA2D1 | c.659–2_659-1insT | NA | Female | 14 | ID | Yes (deeply set eyes, squared facies, bulbous nasal tip, full lips, horizontal eyebrows, enlarged nasal bridge, mouth with downward corners, anteverted nostrils) | Language delay, epilepsy |
| #45 | GPR14 (before UTS2R) | c.844C > T | p.(Gln282 *) | Male | 6 | ID and ASD | Yes (long and large nose, broad nasal bridge, anteverted nostrils, deep philtrum) | Frequent infections |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentino, F.; Bruno, L.P.; Doddato, G.; Giliberti, A.; Tita, R.; Resciniti, S.; Fallerini, C.; Bruttini, M.; Lo Rizzo, C.; Mencarelli, M.A.; et al. Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sci. 2021, 11, 936. https://doi.org/10.3390/brainsci11070936
Valentino F, Bruno LP, Doddato G, Giliberti A, Tita R, Resciniti S, Fallerini C, Bruttini M, Lo Rizzo C, Mencarelli MA, et al. Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sciences. 2021; 11(7):936. https://doi.org/10.3390/brainsci11070936
Chicago/Turabian StyleValentino, Floriana, Lucia Pia Bruno, Gabriella Doddato, Annarita Giliberti, Rossella Tita, Sara Resciniti, Chiara Fallerini, Mirella Bruttini, Caterina Lo Rizzo, Maria Antonietta Mencarelli, and et al. 2021. "Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations" Brain Sciences 11, no. 7: 936. https://doi.org/10.3390/brainsci11070936
APA StyleValentino, F., Bruno, L. P., Doddato, G., Giliberti, A., Tita, R., Resciniti, S., Fallerini, C., Bruttini, M., Lo Rizzo, C., Mencarelli, M. A., Mari, F., Pinto, A. M., Fava, F., Baldassarri, M., Fabbiani, A., Lamacchia, V., Benetti, E., Zguro, K., Furini, S., ... Ariani, F. (2021). Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sciences, 11(7), 936. https://doi.org/10.3390/brainsci11070936