
The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders

Abstract
1. Introduction
2. X-Linked NDD Genes

{kind=link}
| Gene | X-Linked Disorder | Inheritance | Male | Female | Gene Subject to X-Inactivation | References |
|---|---|---|---|---|---|---|
| ABCD1 | Adrenoleukodystrophy/Adrenomyeloneuropathy | Recessive | Death first decade/progressive stiffness and weakness in the legs, development of cognitive and behavioral disturbance beginning in the 2nd decade | unaffected/late onset adrenomyeloneuropathy | Yes | PMID: 23469258 [9]; PMID: 22280810 [10] |
| AFF2 | Intellectual developmental disorder, X-linked 109 | Recessive | Global developmental delay/ID/behavioral dx | mild or unaffected | N.D. | n/a |
| AIFM1 | Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy | Recessive | hypomyelinating leukodystrophy | unaffected carrier females | Yes | PMID: 32337346 [11] |
| ALG13 | Developmental and epileptic encephalopathy 36 | early-onset epileptic encephalopathy, severe intellectual disability | developmental and epileptic encephalopathy-36; unaffected carrier females | Yes | PMID: 28778787 [12] | |
| AP1S2 | PETTIGREW SYNDROME | Recessive | Intellectual disability | unaffected carrier females | ND | n/a |
| ARHGEF9 | Developmental and epileptic encephalopathy 8 | Recessive | profound ID, epilepsy | intellectual disability, unaffected carriers | Yes | PMID: 33600053 [13] |
| ARSE | Chondrodysplasia punctata 1 | Recessive | Developmental delay/ID | not described | ND | n/a |
| ARX | Early infantile epileptic encephalopathyLISX2, Proud syndrome | Recessive | epilepsy and profound ID; brain abnormalities, abnormal genitalia | unaffected; mild phenotype | Yes | PMID: 21416597 [14] |
| ATP6AP2 | Intellectual Disability, X-linked, syndromic, Hedera type | Recessive | ID, epilepsy parkinsonism, spasticity | unaffected | N.D. | n/a |
| ATP7A | Menkes disease | Recessive | epilepsy, developmental delay | unaffected | N.D. | n/a |
| ATRX | Alpha-thalassemia/ID | Dominant | Severe ID and dysmorphic features | mild ID | Yes | PMID: 16100724 [15] |
| ATRX | ATRX ID syndrome | Recessive | severe ID and dysmorphic features | unaffected | Yes | PMID: 16955409 [16] |
| BRWD3 | Intellectual Disability, X-linked 93 | Recessive | ID | unaffected carrier females | N.D. | n/a |
| CASK | Intellectual Disability and microcephaly with pontine and cerebellar hypoplasia | Dominant | ID, microcephaly, pontine, cerebellar hypoplasia | unaffected carrier females, ASD | Yes | PMID: 28944139 [17] |
| CDKL5 | Early infantile epileptic encephalopathy early death | Dominant | milder phenotype, epilepsy and profound ID | severe ID, early onset epilepsy, microcephaly, less severe | Yes | PMID: 24564546 [18] |
| CLIC2 | Intellectual Disability, X-linked, syndromic 32 | Recessive | no affected males | mild learning disabilities | N.D. | n/a |
| CLCN4 | Raynaud-Claes syndrome | Dominant | Sever ID and epilepsy | milder phenotype | Yes | PMID: 27550844 [19] |
| CNKSR2 | Houge type | ND | epilepsy, microcephaly, developmental delay | mild ID, seizure | Yes | PMID: 31414730 [20] |
| CUL4B | Cullun Ring Cabezas type | Recessive | syndromic ID | learning disability | Yes | PMID: 17273978 [21] |
| CXorf56 | CXorf56-Associated ID | ND | moderate ID | generally unaffected or mild phenotype | Yes | PMID: 31822863 [22] |
| DCX | Lissencephaly | ND | ID, epilepsy, brain malformation | mild epilepsy | Yes | PMID: 12838518 [23] |
| DDX3X | Snijders Blok | Recessive | some males with non-syndromic ID | ID, microcephaly | escapes X inactivation | PMID: 30871455 [24] |
| DLG3 | Intellectual Disability, X-linked 90 | Recessive | moderate - severe ID | not affected and affected | Yes | PMID: 28777483 [25] |
| DMD | Duchenne, Muscular dystrophy | Recessive | mild ID | unaffected | Yes | PMID: 27098336 [26] |
| FAM50A | Intellectual developmental disorder, X-linked, syndromic, Armfield type | ID | unaffected | N.D. | n/a | |
| FDG1 | Aarskog–Scott syndrome | Not reported | Facio-genetial dysmorphisms, ADHD, ID | short stature | N.D. | n/a |
| FGF13 | Developmental and epileptic encephalopathy 90 | Dominant/Recessive | epilepsy, developmental delay | epilepsy, developmental delay | N.D. | n/a |
| FMR1 | Fragile X syndrome | Dominant | ID | mild | Yes | PMID: 8825916 [27] |
| FMR1 | Fragile X Tremor Ataxia | Dominant | late onset tremor, ataxia, cognitive decline | FXTAS in 10% of premutation carriers | Yes | PMID: 26609701 [28] |
| FMR1 | Premature Ovarian Failure | n/a | POI in 25% of premutation carriers | N.D. | PMID: 30098699 [29] | |
| FRMPD4 | Intellectual Disability, X-linked 104 | ID | unaffected | N.D. | n/a | |
| FTSJ1 | Intellectual Disability, X-linked 9/44 | Recessive | ID and mood disorder | unaffected carrier females | N.D. | n/a |
| GRIA3 | Intellectual developmental disorder, X-linked, syndromic, Wu type | Recessive | ID | unaffected carrier females | Yes | PMID: 19449417 [30] |
| GPC3/GPC4 | Simpson-Golabi-Behmel | Recessive | ID, congenital malformation | generally unaffected | Yes | PMID: 30048822 [31] |
| HCFC1 | Methylmalonic acidemia | Recessive | ID | not affected | N.D. | n/a |
| HDAC8 | Cornelia de Lange, 5 | Dominant | syndromic ID | mild | Yes | PMID: 22889856 [32] |
| HPRT | Lesch-Nyhan syndrome | Recessive | ID, spastic cerebral palsy and SIB | not affected | Yes | PMID: 6585829 [33] |
| HUWE1 | Intellectual Disability, X-linked | Not reported | moderate -profound syndromic ID | Chiari malformation, ID, dysmorphism | N.D. | n/a |
| IGBP1 | Corpus callosum, agenesis of, with Intellectual Disability, ocular coloboma and micrognathia | Recessive | ID | unaffected | N.D. | n/a |
| IL1RAPL1 | Intellectual Disability, X-linked 21/34 | Recessive | ID, microcephaly | unaffected carrier females | N.D. | n/a |
| IQSEC2 | Intellectual Disability, X-linked 1/78 | Dominant | non-syndromic ID, epilepsy and non-syndromic ID | some with learning disability, milder ID some with epilepsy | escapes X inactivation | PMID: 32564198 [34] |
| KDM5C/JARIDC/SMCX | Claes-Jensen | Recessive | microcephaly, developmental disability | mild phenotype | Yes | [35] |
| KDM6A (UTX) | Kabuki syndrome 2 | Dominant | syndromic ID | similar to males | escapes X inactivation | PMID: 29022598 [7] |
| KLHL15 | Intellectual Disability, X-linked 103 | Recessive | ID, epilepsy, brain malformation | mild or unaffected | Yes | PMID: 24817631 [36] |
| L1CAM | Hydrocephalus. X-linked aqueductal stenosis | Recessive | ID, spastic paraplegia | mild ID, some are not affected | N.D. | n/a |
| LAMP2 | Danon disease | Dominant | ID and myopathy | late onset | escapes X inactivation | PMID: 30871455 [24] |
| MAOA | Monoamine oxidase A def | Recessive | mild ID, behavioral difficulties | not affected | Yes | PMID: 19684479 [37] |
| MECP2 | Rett syndrome | Dominant | early infantile epileptic encephalopathy/death first 2-4 yrs of life | ID, epilepsy, microcephaly, gait and language disorder | Yes | PMID: 18361425 [38]; PMID: 31427717 [39] |
| MECP2 | MECP2 Dup Syndrome | Recessive | profound Intellectual Disability, infantile hypotonia, autistic features, seizures, progressive spasticity, and recurrent infections | mild neuropsychiatric features, such as anxiety. | Yes | PMID: 29141583 [40] |
| MECP2 | PPMX | Recessive | ID, spasticity, tremor, hyperkinetic behavior | unaffected carrier females | N.D. | n/a |
| MED12 (HOPA) | MED12-Related Disorders | Recessive | moderate ID, marfanoid habitus, ID, ptosis, cryptorchidism | unaffected carrier females or Hardikar syndrome | Yes | PMID: 33244166 [41] |
| NEXMIF (KIA2022) | neurite extension & migration | Dominant | severe ID, epilepsy | unaffected, intractable epilepsy and ID | Yes | PMID: 27358180 [42]; PMID: 29717186 [43] |
| NHS | Nance–Horan Syndrome | Dominant | Congenital cataract, microphthalmia, and mild or moderate | mild vision impairment | N.D. | n/a |
| NKAP | Intellectual developmental disorder, X-linked, syndromic, Hackman-Di Donato type | Recessive | ID | unaffected carrier females | N.D. | n/a |
| NLGN3 | Autism risk | ASD | unaffected carrier females | Yes | PMID: 18361425 [38] | |
| NLGN4X | Intellectual Disability, X-linked | no affected males | ID, epilepsy and language disorder | Yes | PMID: 32564284 [44] | |
| NONO | Intellectual Disability, X-linked, syndromic 34 | ID, congenital cardiac malformation | unaffected carrier females | N.D. | n/a | |
| NSDHL | CK syndrome | Recessive | ID, neonatal seizures | N.D. | n/a | |
| OCRL1 | Lowe syndrome | Recessive | ID, cataracts | not affected | skewed X-inactivation | PMID: 7180850 [45] |
| OFD1 | Simpson-Golabi-Behmel | Recessive | early lethality, severe ID | not affected | escapes X inactivation | PMID: 31243241 [46] |
| OFD1 | Joubert 10 | Recessive | ID, congenital malformation | unaffected carrier females | N.D. | n/a |
| OGT | Intellectual Disability, X-linked | Recessive | syndromic ID | not affected | random X-inactivation | PMID: 25136351 [47] |
| OPHN1 | ID, X-linked Congenital Cerebellar hypoplasia | Recessive | ID, hypotonia, ataxia, seizures, macrocephaly, strabismus, dismorphic features | not reported; mild ID, dysmprohic features, strabismus | Yes | PMID: 24105372 [48] |
| PAK3 | Intellectual Disability, X-linked 30/47 | Recessive | ID | unaffected carrier females | N.D. | n/a |
| PCDH19 | Early infantile epileptic encephalopathy, 9 | Not reported | Mosaic males | ID, autism, infantile seizures | Yes | PMID: 22091964 [49] |
| PDHA1 | PDC deficiency | Dominant | early lethality, brain malformation, infantile/childhood onset Leigh mild ataxia | dysmorphism, brain malformation, epilepsy, spastic cp | Yes | PMID: 31673819 [50] |
| PHF6 | Borjeson–Forssman–Lehmann syndrome | Recessive | ID, epilepsy | mild ID | Yes | PMID: 15994862 [51], PMID: 12415272 [52], PMID: 22190899 [53] |
| PHF8 | Syndromic X-linked intellectual disability Siderius type | Recessive | syndromic ID | not affected | Yes | PMID: 18498374 [54] |
| PLP1 | Pelizaeus-Merzbacher Disease | Recessive | leukodystrophy and spastic diplegia | mild or unaffected | Yes | PMID: 10878666 [55]; PMID: 12297985 [56] |
| PORCN | Focal dermal hypoplasia | Dominant | Mosaic males | syndromic ID | Yes | PMID: 17546030 [57]; PMID: 17546031 [58] |
| PQBP1 | Renpenning syndrome | Recessive | ID, microcephaly | unaffected | Yes | PMID: 15811016 [59]; PMID: 31840929 [60] |
| PRPS1 | Arts syndrome | Recessive | ID, ataxia | milder phenotype | Yes | PMID: 24528855 [61] |
| RLIM (RFN12) | Tonne-Kalscheuer syndrome | Not reported | ID, GDD, Autism, congenital malformation | generally unaffected | Yes | PMID: 29728705 [62] |
| RAB39B | Waisman Syndrome | Recessive | ID, epilepsy, Parkinson disease | later onset Parkinson disease and unaffected | N.D. | n/a |
| RPS6KA3 (RSK2) | Coffin-Lowry syndrome | Dominant | syndromic ID, microcephaly | mild ID | Yes | PMID: 12030896 [63] |
| RPS6KA3 (RSK2) | X-Linked MR19 | Dominant | mod ID | mild nonsyndromic ID | N.D. | n/a |
| SLC35A2 | Congenital disorder of glycosylation, type II | Dominant | males are mosaic | Infantile epileptic encephalopathy | Yes | PMID: 24115232 [64] |
| SLC6A8 | Creatine transporter deficiency | Recessive | ID, epilepsy | mild | Yes | PMID: 20528887 [65] |
| SLC9A6 (NHE6) | Christianson syndrome | X-linked | profound ID, epilepsy | ID, learning differences, ADHD, speech delay | Yes | PMID: 18342287 [66] |
| SLC9A7 | Intellectual developmental disorder, X-linked 108 | Recessive | ID | unaffected carrier females | N.D. | n/a |
| SMC1A | DEE85; Cornelia de Lange, 2 | Dominant | Lethal in males; ID, limb malformations, dysmorphic | ID, midline brain defects, seizures | escape X inactivation | PMID: 30871455 [24] |
| SOX3 | Intellectual Disability, X-linked, with isolated growth hormone deficiency | X-linked | ID, panhypopituitarism | unaffected carrier females | N.D. | n/a |
| STAG2 | Holoprosencephaly 13, X-linked | Dominant, Recessive | early lethality, brain malformation, ID | ID, brain malformation/unaffected | N.D. | n/a |
| SYN1 | Epilepsy, X-linked, with variable learning disabilities and behavior disorders | Dominant, Recessive | ID, epilepsy, ASD | epilepsy and ASD | N.D. | n/a |
| SYP | Intellectual Disability, X-linked 96 | Recessive | ID, epilepsy | unaffected | N.D. | n/a |
| TAF1 | XLID 33 | Recessive | syndromic ID | unaffected | Yes | PMID: 26637982 [67] |
| THOC2 | XLID 12/35 | Recessive | mild - moderate ID | not affected | Yes | PMID: 26166480 [68] |
| TSPAN7 | Intellectual Disability, X-linked 58 | Recessive | ID | unaffected carrier females | N.D. | n/a |
| UBE2A | Intellectual Disability, X-linked syndromic, Nascimento-type | Recessive | ID, epilepsy | unaffected carrier females | Yes | PMID: 16909393 [69] |
| UPF3B | XLID 14 | Recessive | Severe non-syndromic ID, Autism | not affected | Yes | PMID: 19238151 [70] |
| USP9X | Syndromic XLID 99 | Dominant | ID, autism, maybe Lethal in males | mild or unaffected or ID, multiple congenital anomalies | escapes X inactivation | PMID: 29022598 [7] |
| USP27X | Intellectual Disability, X-linked 105 | Recessive | ID | unaffected carrier females | N.D. | n/a |
| WDR45 | NBIA5 | Dominant | lethal, mosaics - affected | static encephalopathy, adult onset neurodegeneration, infantile spasms, developmental delay, ID | Yes | PMID: 23176820 [71] |
| ZDHHC9 | Raymond type XLMR | X-linked | non-syndromic ID | unaffected | N.D. | n/a |
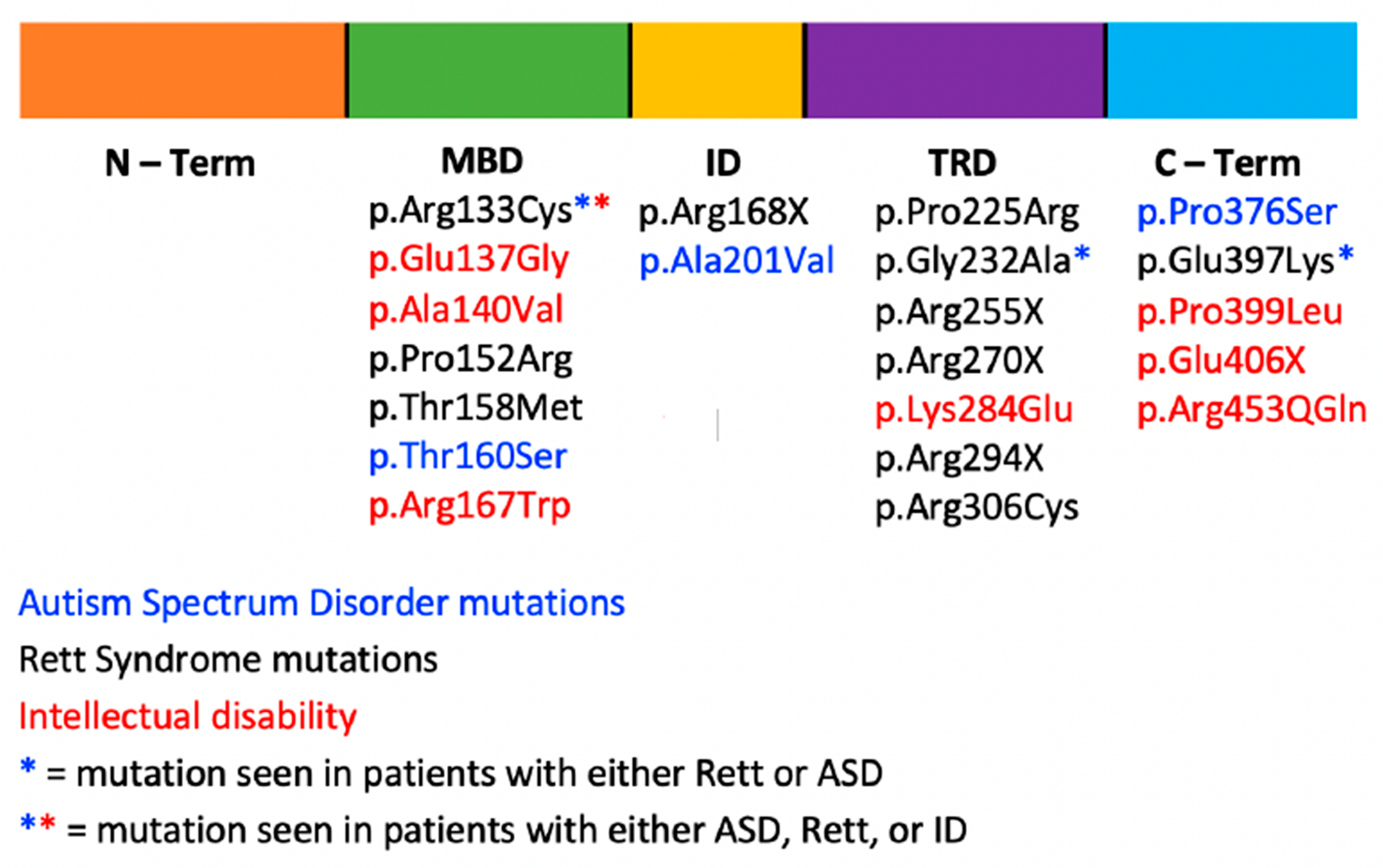
3. MECP2
4. FMR1
4.1. FMR1 Full Mutation
4.2. FMR1 Premutation
4.2.1. Premature Ovarian Insufficiency (POI)
4.2.2. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS)
4.2.3. Children with FMR1 Premutation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laumonnier, F.; Cuthbert, P.C.; Grant, S.G.N. The Role of Neuronal Complexes in Human X-Linked Brain Diseases. Am. J. Hum. Genet. 2007, 80, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Vooren, S.V.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA Sequence of the Human X Chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/ (accessed on 24 April 2021).
- Migeon, B.R. X-Linked Diseases: Susceptible Females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Willard, H.F. X-Inactivation Profile Reveals Extensive Variability in X-Linked Gene Expression in Females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; MacArthur, D.G.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef]
- Salsano, E.; Tabano, S.; Sirchia, S.M.; Colapietro, P.; Castellotti, B.; Gellera, C.; Rimoldi, M.; Pensato, V.; Mariotti, C.; Pareyson, D.; et al. Preferential Expression of Mutant ABCD1 Allele Is Common in Adrenoleukodystrophy Female Carriers but Unrelated to Clinical Symptoms. Orphanet J. Rare Dis. 2012, 7, 10. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, A.; Lin, Y.; Xie, H.; Zhou, C.; Lan, F. Familial Skewed x Chromosome Inactivation in Adrenoleukodystrophy Manifesting Heterozygotes from a Chinese Pedigree. PLoS ONE 2013, 8, e57977. [Google Scholar] [CrossRef]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 Binds to 5hmc Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef]
- Pandolfo, M.; Rai, M.; Remiche, G.; Desmyter, L.; Vandernoot, I. Cerebellar Ataxia, Neuropathy, Hearing Loss, and Intellectual Disability due to AIFM1 Mutation. Neurol. Genet. 2020, 6, e420. [Google Scholar] [CrossRef]
- Hamici, S.; Bastaki, F.; Khalifa, M. Exome Sequence Identified a c.320A > G ALG13 Variant in a Female with Infantile Epileptic Encephalopathy with Normal Glycosylation and Random X Inactivation: Review of the Literature. Eur. J. Med. Genet. 2017, 60, 541–547. [Google Scholar] [CrossRef]
- Ghesh, L.; Besnard, T.; Nizon, M.; Trochu, E.; Landeau-Trottier, G.; Breheret, F.; Thauvin-Robinet, C.; Bruel, A.L.; Kuentz, P.; Coubes, C.; et al. Loss-of-Function Variants in ARHGEF9 are Associated with an X-Linked Intellectual Disability Dominant Disorder. Hum. Mutat. 2021, 42, 498–505. [Google Scholar] [CrossRef]
- Conti, V.; Marini, C.; Gana, S.; Sudi, J.; Dobyns, W.B.; Guerrini, R. Corpus Callosum Agenesis, Severe Mental Retardation, Epilepsy, and Dyskinetic Quadriparesis due to a Novel Mutation in the Homeodomain of ARX. Am. J. Med. Genet. A 2011, 155A, 892–897. [Google Scholar] [CrossRef]
- Wada, T.; Sugie, H.; Fukushima, Y.; Saitoh, S. Non-Skewed X-Inactivation may Cause Mental Retardation in a Female Carrier of X-Linked Alpha-Thalassemia/Mental Retardation Syndrome (ATR-X): X-Inactivation Study of Nine Female Carriers of ATR-X. Am. J. Med. Genet. A 2005, 138, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Badens, C.; Martini, N.; Courrier, S.; DesPortes, V.; Touraine, R.; Levy, N.; Edery, P. ATRX Syndrome in a Girl with a Heterozygous Mutation in the ATRX Zn Finger Domain and a Totally Skewed X-Inactivation Pattern. Am. J. Med. Genet. A 2006, 140, 2212–2215. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Hamazaki, T.; Nishigaki, S.; Kudo, S.; Shintaku, H.; Ondo, Y.; Shimojima, K.; Yamamoto, T. A Novel CASK Mutation Identified in Siblings Exhibiting Developmental Disorders with/without Microcephaly. Intractable Rare Dis Res. 2017, 6, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Bao, X.; Zhang, Q.; Zhang, J.; Cao, G.; Zhang, J.; Li, J.; Wei, L.; Pan, H.; et al. Clinical Features and Gene Mutational Spectrum of CDKL5-Related Diseases in a Cohort of Chinese Patients. BMC Med. Genet. 2014, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.E.; Stuhlmann, T.; Weinert, S.; Haan, E.; Van Esch, H.; Holvoet, M.; Boyle, J.; Leffler, M.; Raynaud, M.; Moraine, C.; et al. De Novo and Inherited Mutations in the X-Linked Gene CLCN4 are Associated with Syndromic Intellectual Disability and Behavior and Seizure Disorders in Males and Females. Mol Psychiatry. 2018, 23, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Polla, D.L.; Saunders, H.R.; de Vries, B.B.A.; van Bokhoven, H.; de Brouwer, A.P.M. A De Novo Variant in the X-Linked Gene CNKSR2 is Associated with Seizures and Mild Intellectual Disability in a Female Patient. Mol. Genet Genomic Med. 2019, 7, e00861. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Q.; Chen, B.; Zhang, X.; Guo, C.; Zhou, H.; Li, J.; Gao, G.; Guo, Y.; Yan, C.; et al. Mutation in CUL4B, which Encodes a Member of Cullin-RING Ubiquitin Ligase Complex, Causes X-Linked Mental Retardation. Am. J. Hum. Genet. 2007, 80, 561–566. [Google Scholar] [CrossRef]
- Rocha, M.E.; Silveira, T.R.D.; Sasaki, E.; Sás, D.M.; Lourenço, C.M.; Kandaswamy, K.K.; Beetz, C.; Rolfs, A.; Bauer, P.; Reardon, W.; et al. Novel Clinical and Genetic Insight into CXorf56-Associated Intellectual Disability. Eur. J. Hum. Genet. 2020, 28, 367–372. [Google Scholar] [CrossRef]
- Guerrini, R.; Moro, F.; Andermann, E.; Hughes, E.; D’Agostino, D.; Carrozzo, R.; Bernasconi, A.; Flinter, F.; Parmeggiani, L.; Volzone, A.; et al. Nonsyndromic Mental Retardation and Cryptogenic Epilepsy in Women with Doublecortin Gene Mutations. Ann. Neurol. 2003, 54, 30–37. [Google Scholar] [CrossRef]
- Wainer Katsir, K.; Linial, M. Human Genes Escaping X-Inactivation Revealed by Single Cell Expression Data. BMC Genom. 2019, 20, 201. [Google Scholar] [CrossRef]
- Gieldon, L.; Mackenroth, L.; Betcheva-Krajcir, E.; Rump, A.; Beck-Wödl, S.; Schallner, J.; Di Donato, N.; Schröck, E.; Tzschach, A. Skewed X-Inactivation in a Family with DLG3-Associated X-Linked Intellectual Disability. Am. J. Med. Genet. A 2017, 173, 2545–2550. [Google Scholar] [CrossRef]
- Viggiano, E.; Ergoli, M.; Picillo, E.; Politano, L. Determining the Role of Skewed X-Chromosome Inactivation in Developing Muscle Symptoms in Carriers of Duchenne Muscular Dystrophy. Hum. Genet. 2016, 135, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kirchgessner, C.U.; Warren, S.T.; Willard, H.F. X Inactivation of the FMR1 Fragile X Mental Retardation Gene. J. Med. Genet. 1995, 32, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Feliu, A.; Badenas, C.; Madrigal, I.; Milà, M. Skewed X Inactivation in Women Carrying the FMR1 Premutation and Its Relation with Fragile-X-Associated Tremor/Ataxia Syndrome. Neurodegener Dis. 2016, 16, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Furtado, C.L.; Luchiari, H.R.; Chielli Pedroso, D.C.; Kogure, G.S.; Caetano, L.C.; Santana, B.A.; Santana, V.P.; Benetti-Pinto, C.L.; Reis, F.M.; Maciel, M.A.; et al. Skewed X-Chromosome Inactivation and Shorter Telomeres Associate with Idiopathic Premature Ovarian Insufficiency. Fertil. Steril. 2018, 110, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Leheup, B.; Béri, M.; Philippe, C.; Grégoire, M.J.; Jonveaux, P. Aberrant GRIA3 Transcripts with Multi-Exon Duplications in a Family with X-Linked Mental Retardation. Am. J. Med. Genet. A 2009, 149A, 1280–1289. [Google Scholar] [CrossRef]
- Schirwani, S.; Novelli, A.; Digilio, M.C.; Bourn, D.; Wilson, V.; Roberts, C.; Dallapiccola, B.; Hobson, E. Duplications of GPC3 and GPC4 Genes in Symptomatic Female Carriers of Simpson-Golabi-Behmel Syndrome type 1. Eur. J. Med. Genet. 2019, 62, 243–247. [Google Scholar] [CrossRef]
- Harakalova, M.; van den Boogaard, M.J.; Sinke, R.; van Lieshout, S.; van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.J.; et al. X-Exome Sequencing Identifies a HDAC8 Variant in a Large Pedigree with X-Linked Intellectual Disability, Truncal Obesity, Gynaecomastia, Hypogonadism and Unusual Face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef]
- Wolf, S.F.; Jolly, D.J.; Lunnen, K.D.; Friedmann, T.; Migeon, B.R. Methylation of the Hypoxanthine Phosphoribosyltransferase Locus on the Human X Chromosome: Implications for X-Chromosome Inactivation. Proc. Natl. Acad. Sci. USA 1984, 81, 2806–2810. [Google Scholar] [CrossRef] [PubMed]
- Wayhelova, M.; Ryzí, M.; Oppelt, J.; Hladilkova, E.; Vallova, V.; Krskova, L.; Vilemova, M.; Polackova, H.; Gaillyova, R.; Kuglik, P. Novel Familial IQSEC2 Pathogenic Sequence Variant Associated with Neurodevelopmental Disorders and Epilepsy. Neurogenetics 2020, 21, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ounap, K.; Puusepp-Benazzouz, H.; Peters, M.; Vaher, U.; Rein, R.; Proos, A.; Field, M.; Reimand, T. A Novel c.2T > C Mutation of the KDM5C/JARID1C Gene in One Large Family with X-Linked Intellectual Disability. Eur. J. Med. Genet. 2012, 55, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Mignon-Ravix, C.; Cacciagli, P.; Choucair, N.; Popovici, C.; Missirian, C.; Milh, M.; Mégarbané, A.; Busa, T.; Julia, S.; Girard, N.; et al. Intragenic Rearrangements in X-Linked Intellectual Deficiency: Results of a-CGH in a Series of 54 Patients and Identification of TRPC5 and KLHL15 as Potential XLID Genes. Am. J. Med. Genet. A 2014, 164A, 1991–1997. [Google Scholar] [CrossRef]
- Stabellini, R.; Vasques, L.R.; de Mello, J.C.; Hernandes, L.M.; Pereira, L.V. MAOA and GYG2 are Submitted to X Chromosome Inactivation in Human Fibroblasts. Epigenetics 2009, 4, 388–393. [Google Scholar] [CrossRef]
- Gong, X.; Bacchelli, E.; Blasi, F.; Toma, C.; Betancur, C.; Chaste, P.; Delorme, R.; Durand, C.M.; Fauchereau, F.; Botros, H.G.; et al. Analysis of X Chromosome Inactivation in Autism Spectrum Disorders. Am. J. Med. Genet. B Neuropsychiatr Genet. 2008, 147B, 830–835. [Google Scholar] [CrossRef]
- Xiol, C.; Vidal, S.; Pascual-Alonso, A.; Blasco, L.; Brandi, N.; Pacheco, P.; Gerotina, E.; O’Callaghan, M.; Pineda, M.; Armstrong, J. X Chromosome Inactivation Does Not Necessarily Determine the Severity of the Phenotype in Rett Syndrome Patients. Sci. Rep. 2019, 9, 11983. [Google Scholar] [CrossRef]
- Li, X.; Xie, H.; Chen, Q.; Yu, X.; Yi, Z.; Li, E.; Zhang, T.; Wang, J.; Zhong, J.; Chen, X. Clinical and Molecular Genetic Characterization of Familial MECP2 Duplication Syndrome in a Chinese Family. BMC Med. Genet. 2017, 18, 131. [Google Scholar] [CrossRef]
- Li, D.; Strong, A.; Shen, K.M.; Cassiman, D.; Van Dyck, M.; Linhares, N.D.; Valadares, E.R.; Wang, T.; Pena, S.D.J.; Jaeken, J.; et al. De Novo Loss-of-Function Variants in X-Linked MED12 are Associated with Hardikar Syndrome in Females. Genet. Med. 2021, 23, 637–644. [Google Scholar] [CrossRef]
- De Lange, I.M.; Helbig, K.L.; Weckhuysen, S.; Møller, R.S.; Velinov, M.; Dolzhanskaya, N.; Marsh, E.; Helbig, I.; Devinsky, O.; Tang, S.; et al. De novo Mutations of KIAA2022 in Females Cause Intellectual Disability and Intractable Epilepsy. J. Med. Genet. 2016, 53, 850–858. [Google Scholar] [CrossRef]
- Lambert, N.; Dauve, C.; Ranza, E.; Makrythanasis, P.; Santoni, F.; Sloan-Béna, F.; Gimelli, S.; Blouin, J.L.; Guipponi, M.; Bottani, A.; et al. Novel NEXMIF Pathogenic Variant in a Boy with Severe Autistic Features, Intellectual Disability, and Epilepsy, and His Mildly Affected Mother. J. Hum. Genet. 2018, 63, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Vianna, E.Q.; Piergiorge, R.M.; Gonçalves, A.P.; Dos Santos, J.M.; Calassara, V.; Rosenberg, C.; Krepischi, A.C.V.; Boy da Silva, R.T.; Dos Santos, S.R.; Ribeiro, M.G.; et al. Understanding the Landscape of X-linked Variants Causing Intellectual Disability in Females Through Extreme X Chromosome Inactivation Skewing. Mol. Neurobiol. 2020, 57, 3671–3684. [Google Scholar] [CrossRef]
- Hittner, H.M.; Carroll, A.J.; Prchal, J.T. Linkage Studies in Carriers of Lowe Oculo-Cerebro-Renal Syndrome. Am. J. Hum. Genet. 1982, 34, 966–971. [Google Scholar]
- Iijima, T.; Hayami, N.; Takaichi, K.; Morisada, N.; Nozu, K.; Iijima, K.; Sawa, N.; Hoshino, J.; Ubara, Y. An Orofaciodigital Syndrome 1 Patient and Her Mother Carry the Same OFD1 Mutation but Have Different X Chromosome Inactivation Patterns. Intern Med. 2019, 58, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Hanover, J.A. X-inactivation Normalizes O-GlcNAc Transferase Levels and Generates an O-GlcNAc-Depleted Barr Body. Front Genet. 2014, 5, 256. [Google Scholar] [CrossRef]
- Santos-Rebouças, C.B.; Belet, S.; Guedes de Almeida, L.; Ribeiro, M.G.; Medina-Acosta, E.; Bahia, P.R.; Alves da Silva, A.F.; Lima dos Santos, F.; Borges de Lacerda, G.C.; Pimentel, M.M.; et al. A Novel in-Frame Deletion Affecting the BAR Domain of OPHN1 in a Family with Intellectual Disability and Hippocampal Alterations. Eur. J. Hum. Genet. 2014, 22, 644–651. [Google Scholar] [CrossRef]
- Vincent, A.K.; Noor, A.; Janson, A.; Minassian, B.A.; Ayub, M.; Vincent, J.B.; Morel, C.F. Identification of Genomic Deletions Spanning the PCDH19 Gene in Two Unrelated Girls with Intellectual Disability and Seizures. Clin. Genet. 2012, 82, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Horga, A.; Woodward, C.E.; Mills, A.; Pareés, I.; Hargreaves, I.P.; Brown, R.M.; Bugiardini, E.; Brooks, T.; Manole, A.; Remzova, E.; et al. Differential Phenotypic Expression of a Novel PDHA1 Mutation in a Female Monozygotic Twin Pair. Hum. Genet. 2019, 138, 1313–1322. [Google Scholar] [CrossRef]
- Crawford, J.; Lower, K.M.; Hennekam, R.C.; Van Esch, H.; Mégarbané, A.; Lynch, S.A.; Turner, G.; Gécz, J. Mutation Screening in Borjeson-Forssman-Lehmann Syndrome: Identification of a Novel De Novo PHF6 Mutation in a Female Patient. J. Med. Genet. 2006, 43, 238–243. [Google Scholar] [CrossRef]
- Lower, K.M.; Turner, G.; Kerr, B.A.; Mathews, K.D.; Shaw, M.A.; Gedeon, A.K.; Schelley, S.; Hoyme, H.E.; White, S.M.; Delatycki, M.B.; et al. Mutations in PHF6 are Associated with Börjeson-Forssman-Lehmann Syndrome. Nat. Genet. 2002, 32, 661–665. [Google Scholar] [CrossRef]
- Berland, S.; Alme, K.; Brendehaug, A.; Houge, G.; Hovland, R. PHF6 Deletions May Cause Borjeson-Forssman-Lehmann Syndrome in Females. Mol. Syndromol. 2011, 1, 294–300. [Google Scholar] [CrossRef]
- Qiao, Y.; Liu, X.; Harvard, C.; Hildebrand, M.J.; Rajcan-Separovic, E.; Holden, J.J.; Lewis, M.E. Autism-Associated Familial Microdeletion of Xp11.22. Clin. Genet. 2008, 74, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Woodward, K.; Kirtland, K.; Dlouhy, S.; Raskind, W.; Bird, T.; Malcolm, S.; Abeliovich, D. X Inactivation Phenotype in Carriers of Pelizaeus-Merzbacher Disease: Skewed in Carriers of a Duplication and Random in Carriers of Point Mutations. Eur. J. Hum. Genet. 2000, 8, 449–454. [Google Scholar] [CrossRef]
- Inoue, K.; Osaka, H.; Thurston, V.C.; Clarke, J.T.; Yoneyama, A.; Rosenbarker, L.; Bird, T.D.; Hodes, M.E.; Shaffer, L.G.; Lupski, J.R. Genomic Rearrangements Resulting in PLP1 Deletion Occur by Nonhomologous End Joining and Cause Different Dysmyelinating Phenotypes in Males and Females. Am. J. Hum. Genet. 2002, 71, 838–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Reid Sutton, V.; Omar Peraza-Llanes, J.; Yu, Z.; Rosetta, R.; Kou, Y.C.; Eble, T.N.; Patel, A.; Thaller, C.; Fang, P.; et al. Mutations in X-linked PORCN, a Putative Regulator of Wnt Signaling, Cause Focal Dermal Hypoplasia. Nat. Genet. 2007, 39, 836–838. [Google Scholar] [CrossRef]
- Grzeschik, K.H.; Bornholdt, D.; Oeffner, F.; König, A.; del Carmen Boente, M.; Enders, H.; Fritz, B.; Hertl, M.; Grasshoff, U.; Höfling, K.; et al. Deficiency of PORCN, a Regulator of Wnt Signaling, is Associated with Focal Dermal Hypoplasia. Nat. Genet. 2007, 39, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Fichera, M.; Falco, M.; Lo Giudice, M.; Castiglia, L.; Guarnaccia, V.; Calì, F.; Spalletta, A.; Scuderi, C.; Avola, E. Skewed X-Inactivation in a Family with Mental Retardation and PQBP1 Gene Mutation. Clin. Genet. 2005, 67, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.Y.; Peñaherrera, M.S.; Du Souich, C.; Huang, L.; Mwenifumbo, J.; Nelson, T.N.; Elliott, A.M.; Adam, S.; CAUSES Study; Eydoux, P.; et al. Renpenning Syndrome in a Female. Am. J. Med. Genet. A 2020, 182, 498–503. [Google Scholar] [CrossRef]
- Synofzik, M.; Müller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wödl, S.; Nabuurs, S.B.; van Kuilenburg, A.B.; de Brouwer, A.P.; Schöls, L. X-Linked Charcot-Marie-Tooth Disease, Arts Syndrome, and Prelingual Non-Syndromic Deafness form a Disease Continuum: Evidence from a Family with a Novel PRPS1 Mutation. Orphanet J. Rare Dis. 2014, 9, 24. [Google Scholar] [CrossRef]
- Frints, S.G.M.; Ozanturk, A.; Rodríguez Criado, G.; Grasshoff, U.; de Hoon, B.; Field, M.; Manouvrier-Hanu, S.; E Hickey, S.; Kammoun, M.; Gripp, K.W.; et al. Pathogenic Variants in E3 Ubiquitin Ligase RLIM/RNF12 Lead to a Syndromic X-Linked Intellectual Disability and Behavior Disorder. Mol. Psychiatry 2019, 24, 1748–1768. [Google Scholar] [CrossRef]
- Simensen, R.J.; Abidi, F.; Collins, J.S.; Schwartz, C.E.; Stevenson, R.E. Cognitive Function in Coffin-Lowry Syndrome. Clin. Genet. 2002, 61, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Nakamura, K.; Osaka, H.; Maegaki, Y.; Haginoya, K.; Mizumoto, S.; Kato, M.; Okamoto, N.; Iai, M.; Kondo, Y.; et al. De Novo Mutations in SLC35A2 Encoding a UDP-Galactose Transporter Cause Early-Onset Epileptic Encephalopathy. Hum. Mutat. 2013, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Van de Kamp, J.M.; Mancini, G.M.; Pouwels, P.J.; Betsalel, O.T.; van Dooren, S.J.; de Koning, I.; Steenweg, M.E.; Jakobs, C.; van der Knaap, M.S.; Salomons, G.S. Clinical Features and X-Inactivation in Females Heterozygous for Creatine Transporter Defect. Clin. Genet. 2011, 79, 264–272. [Google Scholar] [CrossRef]
- Gilfillan, G.D.; Selmer, K.K.; Roxrud, I.; Smith, R.; Kyllerman, M.; Eiklid, K.; Kroken, M.; Mattingsdal, M.; Egeland, T.; Stenmark, H.; et al. SLC9A6 Mutations Cause X-Linked Mental Retardation, Microcephaly, Epilepsy, and Ataxia, a Phenotype Mimicking Angelman Syndrome. Am. J. Hum. Genet. 2008, 82, 1003–1010. [Google Scholar] [CrossRef]
- O’Rawe, J.A.; Wu, Y.; Dörfel, M.J.; Rope, A.F.; Au, P.Y.; Parboosingh, .J.S.; Moon, S.; Kousi, M.; Kosma, K.; Smith, C.S.; et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am. J. Hum. Genet. 2015, 97, 922–932. [Google Scholar] [CrossRef]
- Kumar, R.; Corbett, M.A.; van Bon, B.W.; Woenig, J.A.; Weir, L.; Douglas, E.; Friend, K.L.; Gardner, A.; Shaw, M.; Jolly, L.A.; et al. THOC2 Mutations Implicate mRNA-Export Pathway in X-Linked Intellectual Disability. Am. J. Hum. Genet. 2015, 97, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, R.M.; Otto, P.A.; de Brouwer, A.P.; Vianna-Morgante, A.M. UBE2A, which Encodes a Ubiquitin-Conjugating Enzyme, is Mutated in a Novel X-Linked Mental Retardation Syndrome. Am. J. Hum. Genet. 2006, 79, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B Gene, which Encodes a Protein Widely Expressed in Neurons, are Associated with Nonspecific Mental Retardation with or without Autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome Sequencing Reveals De Novo WDR45 Mutations Causing a Phenotypically Distinct, X-Linked Dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef]
- Srivastava, S.; Sahin, M.; Prock, L. Chapter 22—Translational Medicine Strategies in Drug Development for Neurodevelopmental Disorders. In Handbook of Behavioral Neuroscience; Nomikos, G.G., Feltner, D.E., Eds.; Translational Medicine in CNS Drug Development; Elsevier: Amsterdam, The Netherlands, 2019; Volume 29, pp. 309–331. [Google Scholar]
- Orrico, A.; Lam, C.-W.; Galli, L.; Dotti, M.T.; Hayek, G.; Tong, S.-F.; Poon, P.M.K.; Zappella, M.; Federico, A.; Sorrentino, V. MECP2 Mutation in Male Patients with Non-Specific X-Linked Mental Retardation. FEBS Lett. 2000, 481, 285–288. [Google Scholar] [CrossRef]
- Rett Syndrome. Available online: https://rarediseases.org/rare-diseases/rett-syndrome/ (accessed on 8 June 2020).
- Weaving, L.S.; Williamson, S.L.; Bennetts, B.; Davis, M.; Ellaway, C.J.; Leonard, H.; Thong, M.-K.; Delatycki, M.; Thompson, E.M.; Laing, N.; et al. Effects of MECP2 Mutation Type, Location and X-Inactivation in Modulating Rett Syndrome Phenotype. Am. J. Med. Genet. A 2002, 118A, 103–114. [Google Scholar] [CrossRef]
- Amir, R.E.; Veyver, I.B.V.D.; Schultz, R.; Malicki, D.M.; Tran, C.Q.; Dahle, E.J.; Philippi, A.; Timar, L.; Percy, A.K.; Motil, K.J.; et al. Influence of Mutation Type and X Chromosome Inactivation on Rett Syndrome Phenotypes. Ann. Neurol. 2000, 47, 670–679. [Google Scholar] [CrossRef]
- Cheadle, J.P.; Gill, H.; Fleming, N.; Maynard, J.; Kerr, A.; Leonard, H.; Krawczak, M.; Cooper, D.N.; Lynch, S.; Thomas, N.; et al. Long-Read Sequence Analysis of the MECP2 Gene in Rett Syndrome Patients: Correlation of Disease Severity with Mutation Type and Location. Hum. Mol. Genet. 2000, 9, 1119–1129. [Google Scholar] [CrossRef]
- Weaving, L.; Ellaway, C.; Gecz, J.; Christodoulou, J. Rett Syndrome: Clinical Review and Genetic Update. J. Med. Genet. 2005, 42, 1–7. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, Y.; Bao, X.; Luo, J.; Zhang, X.; Li, J.; Wei, L.; Wu, X. Familial Cases and Male Cases with MECP2 Mutations. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2017, 174, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Jiang, S.; Song, F.; Pan, H.; Li, M.; Wu, X.R. X chromosome inactivation in Rett Syndrome and its correlations with MECP2 mutations and phenotype. J. Child Neurol. 2008, 23, 22–25. [Google Scholar] [CrossRef]
- MECP2 Duplication Syndrome. Available online: https://rarediseases.org/rare-diseases/mecp2-duplication-syndrome/ (accessed on 23 April 2021).
- Schwoerer, J.S.; Laffin, J.; Haun, J.; Raca, G.; Friez, M.J.; Giampietro, P.F. MECP2 Duplication: Possible Cause of Severe Phenotype in Females. Am. J. Med. Genet. A 2014, 164, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 Duplication Syndrome. Am. J. Med. Genet. A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gécz, J.; et al. Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar] [CrossRef]
- Grasshoff, U.; Bonin, M.; Goehring, I.; Ekici, A.; Dufke, A.; Cremer, K.; Wagner, N.; Rossier, E.; Jauch, A.; Walter, M.; et al. De novo MECP2 duplication in two females with random X-inactivation and moderate mental retardation. Eur. J. Hum. Genet. EJHG 2011, 19, 507–512. [Google Scholar] [CrossRef]
- Bijlsma, E.K.; Collins, A.; Papa, F.T.; Tejada, M.I.; Wheeler, P.; Peeters, E.A.; Gijsbers, A.C.; van de Kamp, J.M.; Kriek, M.; Losekoot, M.; et al. Xq28 duplications including MECP2 in five females: Expanding the phenotype to severe mental retardation. Eur. J. Med. Genet. 2012, 55, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Oostra, B.A. Fragile X Syndrome: From Protein Function to Therapy. Am. J. Med. Genet. A 2013, 161, 2809–2821. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X Analysis of 1112 Prenatal Samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef]
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of Fragile X Syndrome: A Systematic Review and Meta-Analysis. Am. J. Med. Genet. A 2014, 164, 1648–1658. [Google Scholar] [CrossRef]
- Hunter, J.E.; Berry-Kravis, E.; Hipp, H.; Todd, P.K. FMR1 Disorders. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Berry-Kravis, E.; Raspa, M.; Loggin-Hester, L.; Bishop, E.; Holiday, D.; Bailey, D.B. Seizures in Fragile X Syndrome: Characteristics and Comorbid Diagnoses. Am. J. Intellect. Dev. Disabil. 2010, 115, 461–472. [Google Scholar] [CrossRef]
- Neri, G. Chapter 1—The Clinical Phenotype of the Fragile X Syndrome and Related Disorders. In Fragile X Syndrome; Willemsen, R., Kooy, R.F., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–16. ISBN 978-0-12-804461-2. [Google Scholar]
- Martínez, R.; Bonilla-Henao, V.; Jimenez, A.; Lucas, M.; Vega, C.; Ramos, I.; Sobrino, F.; Pintado, E. Skewed X Inactivation of the Normal Allele in Fully Mutated Female Carriers Determines the Levels of FMRP in Blood and the Fragile X Phenotype. Mol. Diagn. 2005, 9, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Heine-Suñer, D.; Torres-Juan, L.; Morlà, M.; Busquets, X.; Barceló, F.; Picó, G.; Bonilla, L.; Govea, N.; Bernués, M.; Rosell, J. Fragile-X Syndrome and Skewed X-Chromosome Inactivation within a Family: A Female Member with Complete Inactivation of the Functional X Chromosome. Am. J. Med. Genet. A 2003, 122A, 108–114. [Google Scholar] [CrossRef]
- Martorell, L.; Nascimento, M.T.; Colome, R.; Genovés, J.; Naudó, M.; Nascimento, A. Four Sisters Compound Heterozygotes for the Pre- and Full Mutation in Fragile X Syndrome and a Complete Inactivation of X-Functional Chromosome: Implications for Genetic Counseling. J. Hum. Genet. 2011, 56, 87–90. [Google Scholar] [CrossRef]
- Spath, M.A.; Nillesen, W.N.; Smits, A.P.T.; Feuth, T.B.; Braat, D.D.M.; van Kessel, A.G.; Yntema, H.G. X Chromosome Inactivation Does Not Define the Development of Premature Ovarian Failure in Fragile X Premutation Carriers. Am. J. Med. Genet. A 2010, 152A, 387–393. [Google Scholar] [CrossRef]
- Rodriguez-Revenga, L.; Madrigal, I.; Badenas, C.; Xunclà, M.; Jiménez, L.; Milà, M. Premature Ovarian Failure and Fragile X Female Premutation Carriers: No Evidence for a Skewed X-Chromosome Inactivation Pattern. Menopause 2009, 16, 944–949. [Google Scholar] [CrossRef]
- Johnston-MacAnanny, E.B.; Koty, P.; Pettenati, M.; Brady, M.; Yalcinkaya, T.M.; Schmidt, D.W. The First Case Described: Monozygotic Twin Sisters with the Fragile X Premutation but with a Different Phenotype for Premature Ovarian Failure. Fertil. Steril. 2011, 95, 2431.e13-5. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 Repeat Size with Ovarian Dysfunction. Hum. Reprod. Oxf. Engl. 2005, 20, 402–412. [Google Scholar] [CrossRef]
- Hall, D.A.; Robertson-Dick, E.E.; O’Keefe, J.A.; Hadd, A.G.; Zhou, L.; Berry-Kravis, E. X-Inactivation in the Clinical Phenotype of Fragile X Premutation Carrier Sisters. Neurol. Genet. 2016, 2, e45. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-Associated Tremor/Ataxia Syndrome in Sisters Related to X-Inactivation. Ann. Neurol. 2005, 57, 144–147. [Google Scholar] [CrossRef]
- Bailey, D.B.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-Occurring Conditions Associated with FMR1 Gene Variations: Findings from a National Parent Survey. Am. J. Med. Genet. A 2008, 146A, 2060–2069. [Google Scholar] [CrossRef]
- Renda, M.M.; Voigt, R.G.; Babovic-Vuksanovic, D.; Highsmith, W.E.; Vinson, S.S.; Sadowski, C.M.; Hagerman, R.J. Neurodevelopmental Disabilities in Children with Intermediate and Premutation Range Fragile X Cytosine-Guanine-Guanine Expansions. J. Child Neurol. 2014, 29, 326–330. [Google Scholar] [CrossRef]
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism Spectrum Disorders and Attention-Deficit/Hyperactivity Disorder in Boys with the Fragile X Premutation. J. Dev. Behav. Pediatr. 2006, 27, S137. [Google Scholar] [CrossRef] [PubMed]
- Myers, G.F.; Mazzocco, M.M.M.; Maddalena, A.; Reiss, A.L. No Widespread Psychological Effect of the Fragile X Premutation in Childhood: Evidence from a Preliminary Controlled Study. J. Dev. Behav. Pediatr. 2001, 22, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Morleo, M.; Franco, B. Microphthalmia with Linear Skin Defects Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Carrette, L.L.G.; Wang, C.-Y.; Wei, C.; Press, W.; Ma, W.; Kelleher, R.J.; Lee, J.T. A Mixed Modality Approach towards Xi Reactivation for Rett Syndrome and Other X-Linked Disorders. Proc. Natl. Acad. Sci. USA 2018, 115, E668–E675. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brand, B.A.; Blesson, A.E.; Smith-Hicks, C.L. The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders. Brain Sci. 2021, 11, 904. https://doi.org/10.3390/brainsci11070904
Brand BA, Blesson AE, Smith-Hicks CL. The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders. Brain Sciences. 2021; 11(7):904. https://doi.org/10.3390/brainsci11070904
Chicago/Turabian StyleBrand, Boudewien A, Alyssa E Blesson, and Constance L. Smith-Hicks. 2021. "The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders" Brain Sciences 11, no. 7: 904. https://doi.org/10.3390/brainsci11070904
APA StyleBrand, B. A., Blesson, A. E., & Smith-Hicks, C. L. (2021). The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders. Brain Sciences, 11(7), 904. https://doi.org/10.3390/brainsci11070904