
Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
- The age at onset, diagnosis, and death, or age of living patients as of 31 December 2020,
- The family history of neuropathy or ATTR amyloidosis,
- The type of TTR mutation,
- The clinical and neurological examination at diagnosis,
- Electromyography and nerve conduction studies at diagnosis,
- The presence of autonomic nervous system involvement at diagnosis, when available and based on autonomic cardiovascular tests, or, in their absence, based on clinical evaluation,
- The presence of cardiac involvement at diagnosis based on ECG, echocardiogram, atrial natriuretic hormone dosage (nt-pro-BNP), and, in some cases, 99 m Tc-3, 3-diphosphono-1, 2-propanodicarboxylic acid scintigraphy or cardiac magnetic resonance (CMR) with T2-weighted imaging and late gadolinium enhancement,
- Sural nerve biopsy in selected cases,
- Therapeutic drugs (or procedures) and the overall duration of their administration,
- Symptomatic treatment.
Statistical Analysis
3. Results
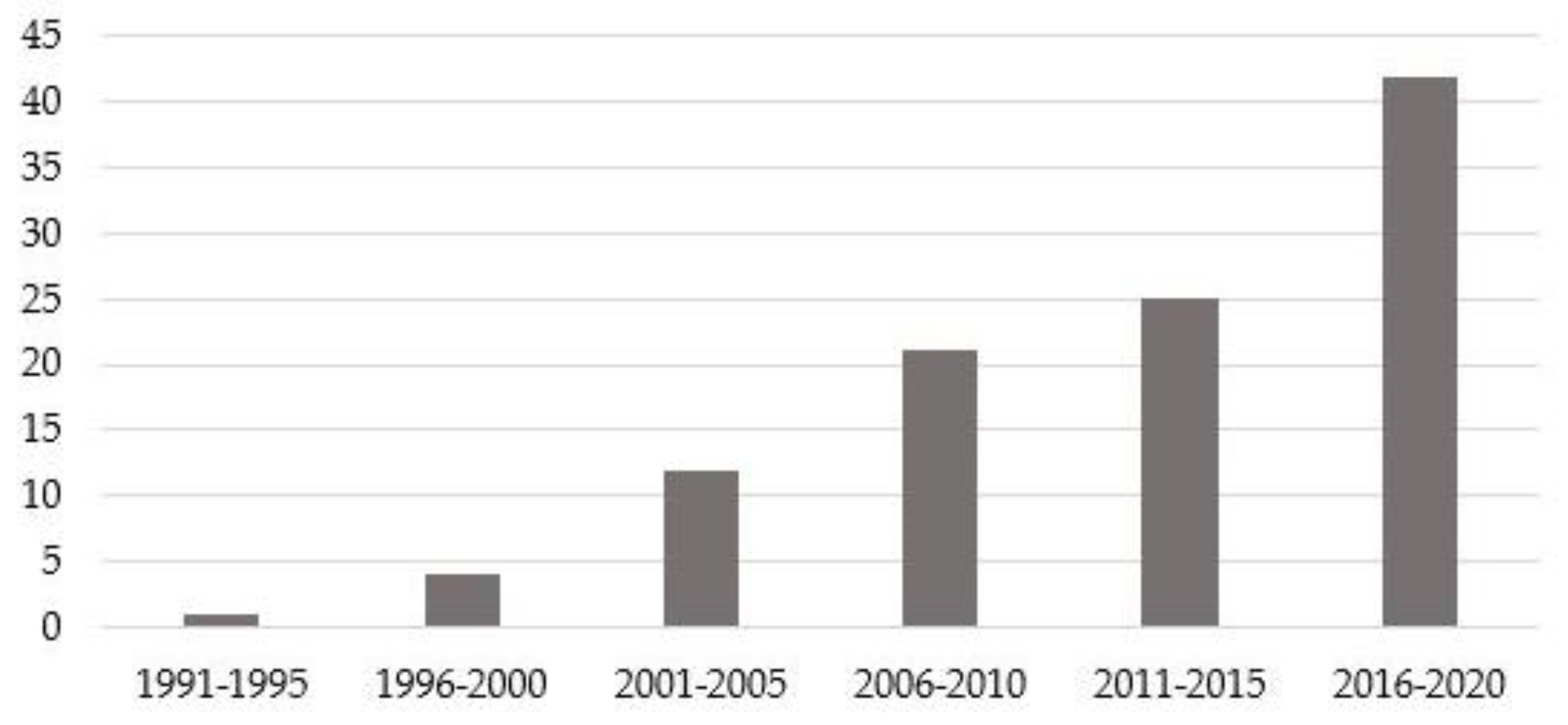
3.1. General Aspects
3.2. Actual Scenario
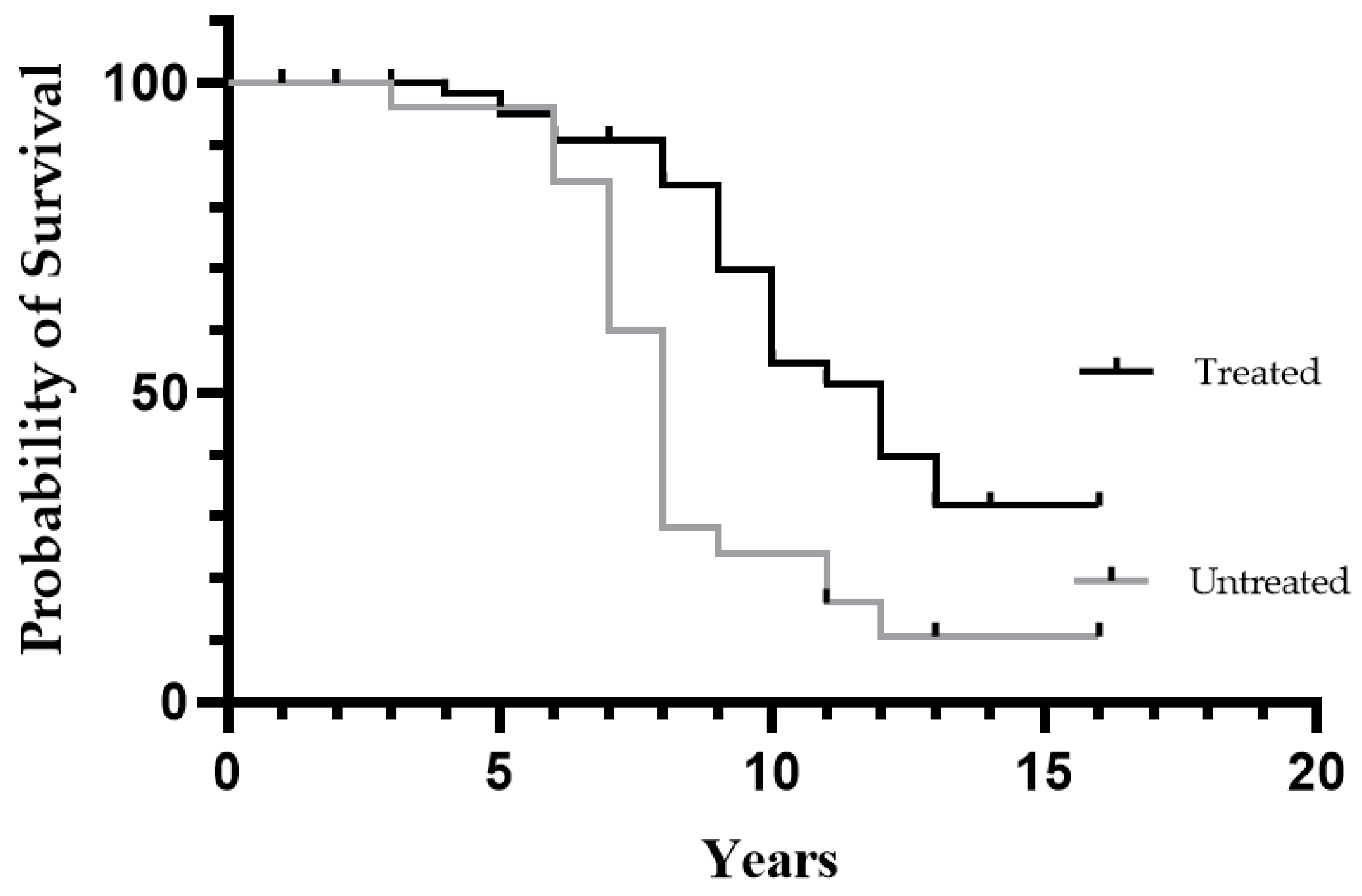
3.3. Comparison of Treated and Untreated Patients
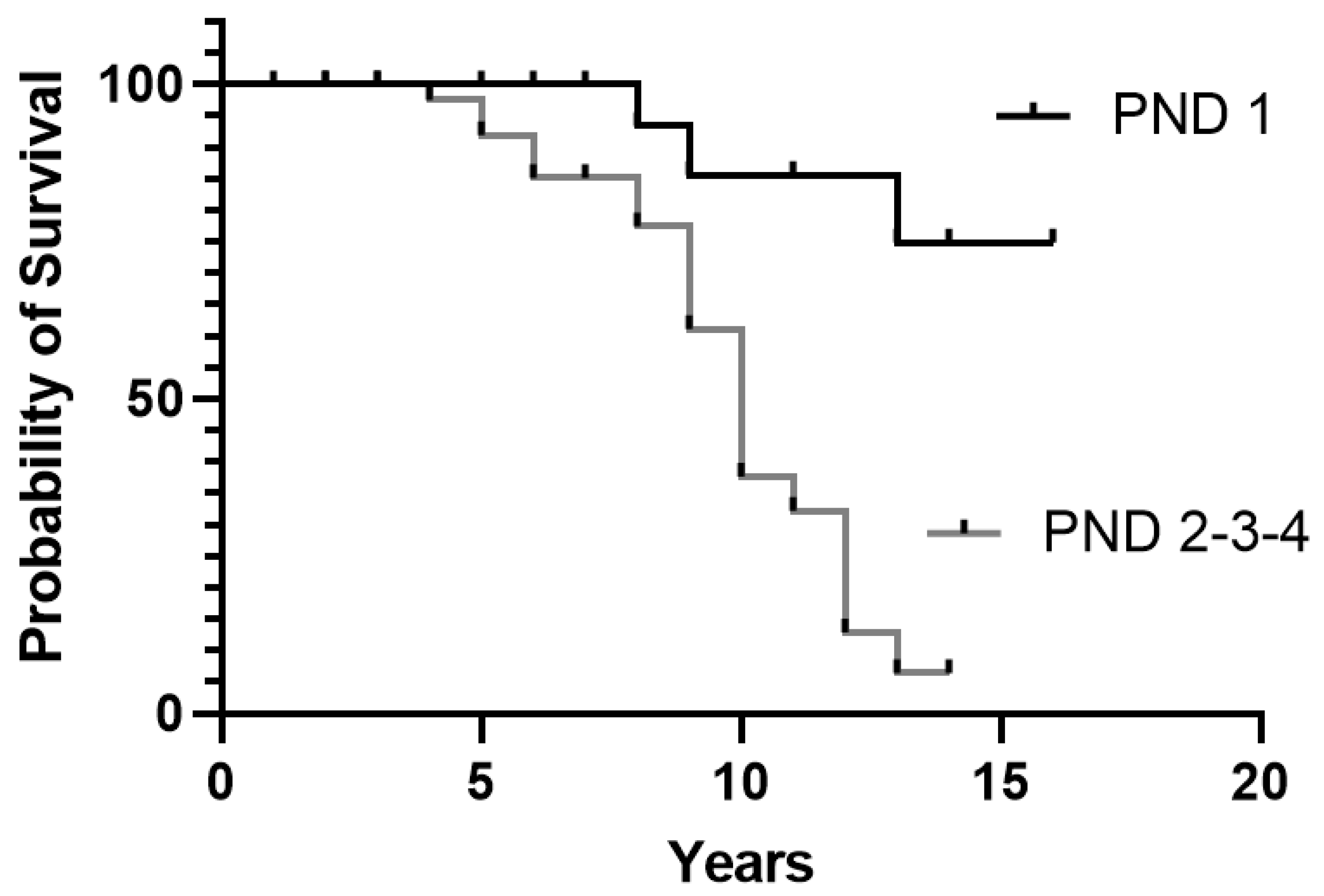
3.4. Relation between Clinical Characteristics at the Onset and Survival in Treated Patients
3.5. Symptomatic Drugs
4. Discussion
4.1. Epidemiological Aspect
4.2. Diagnostic Delay and Carpal Tunnel Syndrome
4.3. Treatment Used
4.4. Effect on Survival
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert Consensus Recommendations to Improve Diagnosis of ATTR Amyloidosis with Polyneuropathy. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the Global Prevalence of Transthyretin Familial Amyloid Polyneuropathy: ATTR-FAP Global Prevalence. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Waddington-Cruz, M.; Schmidt, H.; Botteman, M.F.; Carter, J.A.; Stewart, M.; Hopps, M.; Fallet, S.; Amass, L. Epidemiological and Clinical Characteristics of Symptomatic Hereditary Transthyretin Amyloid Polyneuropathy: A Global Case Series. Orphanet J. Rare Dis. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Obici, L.; Bartolomei, I.; Cappelli, F.; Luigetti, M.; Fenu, S.; Cavallaro, T.; Chiappini, M.G.; Gemelli, C.; Pradotto, L.G.; et al. ATTRv Amyloidosis Italian Registry: Clinical and Epidemiological Data. Amyloid 2020, 27, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Parman, Y.; Adams, D.; Obici, L.; Galán, L.; Guergueltcheva, V.; Suhr, O.B.; Coelho, T.; European Network for TTR-FAP (ATTReuNET). Sixty Years of Transthyretin Familial Amyloid Polyneuropathy (TTR-FAP) in Europe: Where Are We Now? A European Network Approach to Defining the Epidemiology and Management Patterns for TTR-FAP. Curr. Opin. Neurol. 2016, 29 (Suppl. 1), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Sobue, G. Late-Onset Familial Amyloid Polyneuropathy in Japan. Amyloid 2012, 19 (Suppl. 1), 55–57. [Google Scholar] [CrossRef]
- Mariani, L.-L.; Lozeron, P.; Théaudin, M.; Mincheva, Z.; Signate, A.; Ducot, B.; Algalarrondo, V.; Denier, C.; Adam, C.; Nicolas, G.; et al. Genotype-Phenotype Correlation and Course of Transthyretin Familial Amyloid Polyneuropathies in France. Ann. Neurol. 2015, 78, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Durmuş-Tekçe, H.; Matur, Z.; Mert Atmaca, M.; Poda, M.; Çakar, A.; Hıdır Ulaş, Ü.; Oflazer-Serdaroğlu, P.; Deymeer, F.; Parman, Y.G. Genotypic and Phenotypic Presentation of Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP) in Turkey. Neuromuscul. Disord. 2016, 26, 441–446. [Google Scholar] [CrossRef]
- Russo, M.; Mazzeo, A.; Stancanelli, C.; Di Leo, R.; Gentile, L.; Di Bella, G.; Minutoli, F.; Baldari, S.; Vita, G. Transthyretin-Related Familial Amyloidotic Polyneuropathy: Description of a Cohort of Patients with Leu64 Mutation and Late Onset. J. Peripher. Nerv. Syst. 2012, 17, 385–390. [Google Scholar] [CrossRef]
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancanelli, C.; Gentile, L.; Baldari, S.; Carerj, S.; Toscano, A.; Vita, G. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J. Neuromuscul. Dis. 2015, 2, S39–S48. [Google Scholar] [CrossRef]
- Law, S.; Petrie, A.; Chacko, L.; Cohen, O.C.; Ravichandran, S.; Gilbertson, J.A.; Rowczenio, D.; Wechalekar, A.; Martinez-Naharro, A.; Lachmann, H.J.; et al. Disease Progression in Cardiac Transthyretin Amyloidosis Is Indicated by Serial Calculation of National Amyloidosis Centre Transthyretin Amyloidosis Stage. ESC Heart Fail. 2020. [Google Scholar] [CrossRef] [PubMed]
- Foss, T.R.; Wiseman, R.L.; Kelly, J.W. The Pathway by Which the Tetrameric Protein Transthyretin Dissociates. Biochemistry 2005, 44, 15525–15533. [Google Scholar] [CrossRef]
- Blake, C.C.F.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of Prealbumin: Secondary, Tertiary and Quaternary Interactions Determined by Fourier Refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Monaco, H.; Rizzi, M.; Coda, A. Structure of a Complex of Two Plasma Proteins: Transthyretin and Retinol-Binding Protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Russo, M.; Gentile, L.; Toscano, A.; Aguennouz, M.; Vita, G.; Mazzeo, A. Advances in Treatment of ATTRv Amyloidosis: State of the Art and Future Prospects. Brain Sci. 2020, 10, 952. [Google Scholar] [CrossRef] [PubMed]
- Vita, G.; Vita, G.L.; Stancanelli, C.; Gentile, L.; Russo, M.; Mazzeo, A. Genetic Neuromuscular Disorders: Living the Era of a Therapeutic Revolution. Part 1: Peripheral Neuropathies. Neurol. Sci. 2019, 40, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Plante-Bordeneuve, V. Transthyretin Familial Amyloid Polyneuropathy: An Update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary Transthyretin Amyloidosis: A Model of Medical Progress for a Fatal Disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Gagliardi, C.; Perfetto, F.; Lorenzini, M.; Ferlini, A.; Salvi, F.; Milandri, A.; Quarta, C.C.; Taborchi, G.; Bartolini, S.; Frusconi, S.; et al. Phenotypic Profile of Ile68Leu Transthyretin Amyloidosis: An Underdiagnosed Cause of Heart Failure. Eur. J. Heart Fail. 2018, 20, 1417–1425. [Google Scholar] [CrossRef]
- Gentile, L.; Di Bella, G.; Minutoli, F.; Cucinotta, F.; Obici, L.; Mussinelli, R.; Arimatea, I.; Russo, M.; Toscano, A.; Vita, G.; et al. Description of a Large Cohort of Caucasian Patients with V122I ATTRv Amyloidosis: Neurological and Cardiological Features. J. Peripher. Nerv. Syst. 2020, 25, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Vita, G.L.; Stancanelli, C.; Mazzeo, A.; Vita, G.; Messina, S. Parenteral Nutrition Improves Nutritional Status, Autonomic Symptoms and Quality of Life in Transthyretin Amyloid Polyneuropathy. Neuromuscul. Disord. 2016, 26, 374–377. [Google Scholar] [CrossRef]
- Vita, G.L.; Aguennouz, M.; Polito, F.; Oteri, R.; Russo, M.; Gentile, L.; Barbagallo, C.; Ragusa, M.; Rodolico, C.; Di Giorgio, R.M.; et al. Circulating MicroRNAs Profile in Patients With Transthyretin Variant Amyloidosis. Front. Mol. Neurosci. 2020, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N. Treatment of Hereditary and Acquired Forms of Transthyretin Amyloidosis in the Era of Personalized Medicine: The Role of Randomized Controlled Trials. Amyloid 2019, 26, 55–65. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.-J.; et al. Tafamidis for Transthyretin Familial Amyloid Polyneuropathy: A Randomized, Controlled Trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Cortese, A.; Vita, G.; Luigetti, M.; Russo, M.; Bisogni, G.; Sabatelli, M.; Manganelli, F.; Santoro, L.; Cavallaro, T.; Fabrizi, G.M.; et al. Monitoring Effectiveness and Safety of Tafamidis in Transthyretin Amyloidosis in Italy: A Longitudinal Multicenter Study in a Non-Endemic Area. J. Neurol. 2016, 263, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.N.; Hund, E.; Purrucker, J.C.; Huber, L.; Kimmich, C.; Aus dem Siepen, F.; Hein, S.; Kristen, A.V.; Hinderhofer, K.; Kollmer, J.; et al. Real-World Outcomes in Non-Endemic Hereditary Transthyretin Amyloidosis with Polyneuropathy: A 20-Year German Single-Referral Centre Experience. Amyloid 2020, 1–9. [Google Scholar] [CrossRef]
- Coelho, T.; Inês, M.; Conceição, I.; Soares, M.; de Carvalho, M.; Costa, J. Natural History and Survival in Stage 1 Val30Met Transthyretin Familial Amyloid Polyneuropathy. Neurology 2018, 91, e1999–e2009. [Google Scholar] [CrossRef]
- Merlini, G.; Coelho, T.; Waddington Cruz, M.; Li, H.; Stewart, M.; Ebede, B. Evaluation of Mortality during Long-Term Treatment with Tafamidis for Transthyretin Amyloidosis with Polyneuropathy: Clinical Trial Results up to 8.5 Years. Neurol. Ther. 2020, 9, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Denier, C.; Ducot, B.; Husson, H.; Lozeron, P.; Adams, D.; Meyer, L.; Said, G.; Planté-Bordeneuve, V. A Brief Compound Test for Assessment of Autonomic and Sensory-Motor Dysfunction in Familial Amyloid Polyneuropathy. J. Neurol. 2007, 254, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Adams, D. Recent Advances in the Treatment of Familial Amyloid Polyneuropathy. Ther. Adv. Neurol. Disord. 2013, 6, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Quarta, C.C.; Obici, L.; Perfetto, F.; Longhi, S.; Salvi, F.; Biagini, E.; Lorenzini, M.; Grigioni, F.; Leone, O.; et al. Disease Profile and Differential Diagnosis of Hereditary Transthyretin-Related Amyloidosis with Exclusively Cardiac Phenotype: An Italian Perspective. Eur. Heart J. 2013, 34, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Stancanelli, C.; Gentile, L.; Di Bella, G.; Minutoli, F.; Russo, M.; Vita, G.; Mazzeo, A. Phenotypic Variability of TTR Val122Ile Mutation: A Caucasian Patient with Axonal Neuropathy and Normal Heart. Neurol. Sci. 2017, 38, 525–526. [Google Scholar] [CrossRef]
- Milandri, A.; Farioli, A.; Gagliardi, C.; Longhi, S.; Salvi, F.; Curti, S.; Foffi, S.; Caponetti, A.G.; Lorenzini, M.; Ferlini, A.; et al. Carpal Tunnel Syndrome in Cardiac Amyloidosis: Implications for Early Diagnosis and Prognostic Role across the Spectrum of Aetiologies. Eur. J. Heart Fail. 2020, 22, 507–515. [Google Scholar] [CrossRef]
- Samões, R.; Taipa, R.; Valdrez, K.; Gonçalves, I.; Melo Pires, M.; Martins da Silva, A.; Coelho, T. Amyloid Detection in the Transverse Carpal Ligament of Patients with Hereditary ATTR V30M Amyloidosis and Carpal Tunnel Syndrome. Amyloid 2017, 24, 73–77. [Google Scholar] [CrossRef]
- Minutoli, F.; Di Bella, G.; Mazzeo, A.; Laudicella, R.; Gentile, L.; Russo, M.; Vita, G.; Baldari, S. Serial Scanning with 99mTc-3, 3-Diphosphono-1, 2-Propanodicarboxylic Acid (99mTc-DPD) for Early Detection of Cardiac Amyloid Deposition and Prediction of Clinical Worsening in Subjects Carrying a Transthyretin Gene Mutation. J. Nucl. Cardiol. 2019. [Google Scholar] [CrossRef]
- Minutoli, F.; Di Bella, G.; Mazzeo, A.; Donato, R.; Russo, M.; Scribano, E.; Baldari, S. Comparison between (99m)Tc-Diphosphonate Imaging and MRI with Late Gadolinium Enhancement in Evaluating Cardiac Involvement in Patients with Transthyretin Familial Amyloid Polyneuropathy. AJR Am. J. Roentgenol. 2013, 200, W256–W265. [Google Scholar] [CrossRef]
- Di Bella, G.; Minutoli, F.; Madaffari, A.; Mazzeo, A.; Russo, M.; Donato, R.; Zito, C.; Aquaro, G.D.; Piccione, M.C.; Pedri, S.; et al. Left Atrial Function in Cardiac Amyloidosis. J. Cardiovasc. Med. 2016, 17, 113–121. [Google Scholar] [CrossRef]
- Said, G.; Grippon, S.; Kirkpatrick, P. Tafamidis. Nat. Rev. Drug Discov. 2012, 11, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Aimo, A.; Emdin, M. Tafamidis Is Entering the Clinical Arena for the Treatment of Transthyretin-Related Cardiomyopathy: Certainties and Unmet Needs. Eur. J. Heart Fail. 2021. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Lin, H.; Gollob, J.; Agarwal, S.; Betts, M.; Fahrbach, K.; Chitnis, M.; Polydefkis, M. An Indirect Treatment Comparison of the Efficacy of Patisiran and Tafamidis for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis with Polyneuropathy. Expert Opin. Pharmacother. 2019, 20, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Suhr, O.B.; Conceiçao, I.; Schmidt, H.H.-J.; Trigo, P.; Kelly, J.W.; et al. Long-Term Effects of Tafamidis for the Treatment of Transthyretin Familial Amyloid Polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Polydefkis, M.; González-Duarte, A.; Wixner, J.; Kristen, A.V.; Schmidt, H.H.; Berk, J.L.; Losada López, I.A.; Dispenzieri, A.; Quan, D.; et al. Long-Term Safety and Efficacy of Patisiran for Hereditary Transthyretin-Mediated Amyloidosis with Polyneuropathy: 12-Month Results of an Open-Label Extension Study. Lancet Neurol. 2021, 20, 49–59. [Google Scholar] [CrossRef]
- Okamoto, S.; Wixner, J.; Obayashi, K.; Ando, Y.; Ericzon, B.-G.; Friman, S.; Uchino, M.; Suhr, O.B. Liver Transplantation for Familial Amyloidotic Polyneuropathy: Impact on Swedish Patients’ Survival. Liver Transplant. 2009, 15, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Ericzon, B.-G.; Friman, S. Long-Term Follow-up of Survival of Liver Transplant Recipients with Familial Amyloid Polyneuropathy (Portuguese Type). Liver Transplant. 2002, 8, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Ericzon, B.-G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R.; Furtado, E.; Barroso, E.; Daniel, J.; Samuel, D.; et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef]
- Liepnieks, J.J.; Zhang, L.Q.; Benson, M.D. Progression of Transthyretin Amyloid Neuropathy after Liver Transplantation. Neurology 2010, 75, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Chung, K.; Lee, J.H.; Cohn, W.; Whitelegge, J.P.; Benson, M.D.; Eisenberg, D.S. Amyloid Seeding of Transthyretin by Ex Vivo Cardiac Fibrils and Its Inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E6741–E6750. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Nguyen, B.A.; Chung, K.; Wang, Y.; Ortega, A.; Lee, J.H.; Coelho, T.; Bijzet, J.; Benson, M.D.; Eisenberg, D.S. A Pair of Peptides Inhibits Seeding of the Hormone Transporter Transthyretin into Amyloid Fibrils. J. Biol. Chem. 2019, 294, 6130–6141. [Google Scholar] [CrossRef] [PubMed]
- Emdin, M.; Aimo, A.; Rapezzi, C.; Fontana, M.; Perfetto, F.; Seferović, P.M.; Barison, A.; Castiglione, V.; Vergaro, G.; Giannoni, A.; et al. Treatment of Cardiac Transthyretin Amyloidosis: An Update. Eur. Heart J. 2019, 40, 3699–3706. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Total | F64L | E89Q | V122I | T49A | V30M | |
|---|---|---|---|---|---|---|
| Patients, n (m/f) | 105 (68/37) | 51 (36/15) | 38 (21/17) | 6 (6/0) | 8 (3/5) | 2 (2/0) |
| Age at onset, mean (range) | 59.3 (33.0–81.2) | 66.6 (46.3–81.2) | 51.7 (36.4–76.2) | 65.3 (62.3–69.1) | 43.6 (33.0–56.3) | 62.4 (53.7–71.1) |
| Age at diagnosis, mean (range) | 62.2 (33.9–85.3) | 70.1 (52.3–85.3) | 53.5 (40.2–74.8) | 70.1 (66.0–76.1) | 45.4 (33.9–57.7) | 65.9 (56.4–75.3) |
| Diagnostic delay (years), mean (range) | 2.8 (0–16) | 3.5 (0–16) | 1.9 (0–6) | 4.8 (3–7) | 1.75 (0–4) | 3.5 (3–4) |
| Probands, n (%) | 50 (47.6) | 35 (68.6) | 7 (18.4) | 1 (16.7) | 1 (12.5) | 0 |
| Late onset, n (%) | 81 (77.1) | 50 (98.0) | 22 (57.8) | 6 (100) | 1 (12.5) | 2 (100) |
| Neurologic symptoms at diagnosis, n (%) | 103 (98.0) | 51 (100) | 36 (94.7) | 6 (100) | 8 (100) | 2 (100) |
| Sensory | 97 (92.4) | 45 (88.2) | 36 (94.7) | 6 (100) | 8 (100) | 2 (100) |
| Motor | 60 (57.1) | 37 (72.5) | 15 (39.5) | 0 | 6 (75) | 2 (100) |
| Gait difficulties | 46 (43.8) | 32 (62.7) | 11 (28.9) | 0 | 1 (12.5) | 2 (100) |
| CTS | 62 (59.0) | 30 (58.8) | 24 (63.2) | 4 (66.7) | 3 (37.5) | 1 (50) |
| Cardiac involvement at diagnosis, n (%) | 48 (45.7) | 9 (17.6) | 31 (81.6) | 5 (83.3) | 3 (37.5) | 0 |
| Dysautonomia at diagnosis, n (%) | 60 (57.1) | 24 (47.1) | 26 (68.4) | 1 (16.7) | 8 (100) | 1 (50) |
| CADT score point loss | 5.5 | 6.2 | 5.1 | 6 | 5.2 | 2 |
| Weight loss, n (%) | 53 (50.5) | 25 (49.0) | 16 (42.1) | 1 (50) | 5 (87.5) | 2 (100) |
| Living patients, n (%) | 59 (56.1) | 27 (52.9) | 23 (60.5) | 3 (50) | 4 (50) | 2 (100) |
| Disease duration in living pt. (yrs) | 6.15 (0.4–16) | 4.8 (0.4–16) | 6.8 (0.5–13.6) | 8.9 (4.9–13.6) | 7.3 (5.5–8.6) | 9.6 (5.5–13.7) |
| Deceased patients, n (%) | 46 (43.8) | 24 (47.0) | 15 (39.4) | 2 (33.3) | 5 (62.5) | 0 |
| Disease duration in deceased pt. (yrs) | 8.4 (3.1–13.0) | 8.5 (4.0–11.8) | 8.2 (3.1–13.0) | 7.5 (6.3–8.7) | 9.2 (7.0–12.9) | N.A. |
| Treated | Untreated | p | ||
|---|---|---|---|---|
| Patients, n (m/f) | 71 (51/20) | 34 (16/18) | ||
| Age at onset, mean (range) | 58.5 (33.0–81.2) | 61.0 (37.5–75.7) | 0.24 | |
| Diagnosis before 2011, n (%) | 17 (43) | 23 (57) | ||
| Diagnosis after 2011, n (%) | 54 (83) | 11 (17) | ||
| Type of mutation: F64L | 30 | 21 | ||
| E89Q | 28 | 10 | ||
| T49A | 6 | 2 | ||
| V122I | 5 | 1 | ||
| V30M | 2 | 0 | ||
| Neurologic symptoms at diagnosis, n (%) | 69 (97) | 34 (100) | ||
| Cardiac involvement at diagnosis, n (%) | 37 (52) | 11 (32) | ||
| Dysautonomia at diagnosis, n (%) | 43 (60) | 17 (50) | ||
| Weight loss at diagnosis, n (%) | 39 (55) | 14 (41) | ||
| Years of disease, total (mean) | 531 (7.48) | 220 (6.47) | ||
| Years of treatment received (n patients/mean) | 299 | (71/4.2) | ||
| Tafamidis | 192 | (52/3.7) | ||
| Liver transplantation | 52 | (6/8.7) | ||
| Patisiran | 37 | (20/1.9) | ||
| Inotersen | 14 | (7/2) | ||
| Diflunisal | 4 | (2/2) | ||
| Diagnostic delay, total (mean) | 180 (2.5) | 119 (3.5) | ||
| Time out of treatment for other reasons | 52 | 101 | ||
| Deceased patients, n (%) | 24 (34) | 22 (65) | ||
| Disease duration in deceased patients, mean (range) | 9.1 (4–13) | 7.7 (3–12) | 0.04 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, M.; Gentile, L.; Di Stefano, V.; Di Bella, G.; Minutoli, F.; Toscano, A.; Brighina, F.; Vita, G.; Mazzeo, A. Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area. Brain Sci. 2021, 11, 545. https://doi.org/10.3390/brainsci11050545
Russo M, Gentile L, Di Stefano V, Di Bella G, Minutoli F, Toscano A, Brighina F, Vita G, Mazzeo A. Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area. Brain Sciences. 2021; 11(5):545. https://doi.org/10.3390/brainsci11050545
Chicago/Turabian StyleRusso, Massimo, Luca Gentile, Vincenzo Di Stefano, Gianluca Di Bella, Fabio Minutoli, Antonio Toscano, Filippo Brighina, Giuseppe Vita, and Anna Mazzeo. 2021. "Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area" Brain Sciences 11, no. 5: 545. https://doi.org/10.3390/brainsci11050545
APA StyleRusso, M., Gentile, L., Di Stefano, V., Di Bella, G., Minutoli, F., Toscano, A., Brighina, F., Vita, G., & Mazzeo, A. (2021). Use of Drugs for ATTRv Amyloidosis in the Real World: How Therapy Is Changing Survival in a Non-Endemic Area. Brain Sciences, 11(5), 545. https://doi.org/10.3390/brainsci11050545