
Protective Effects of PGC-1α Activators on Ischemic Stroke in a Rat Model of Photochemically Induced Thrombosis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Focal Ischemic Stroke
2.3. Study Design and Drug Treatment
2.4. Estimation of the Infarcted Core Area and the Size/Cell Profile of Ischemic Penumbra
2.5. Western Blot Analysis
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
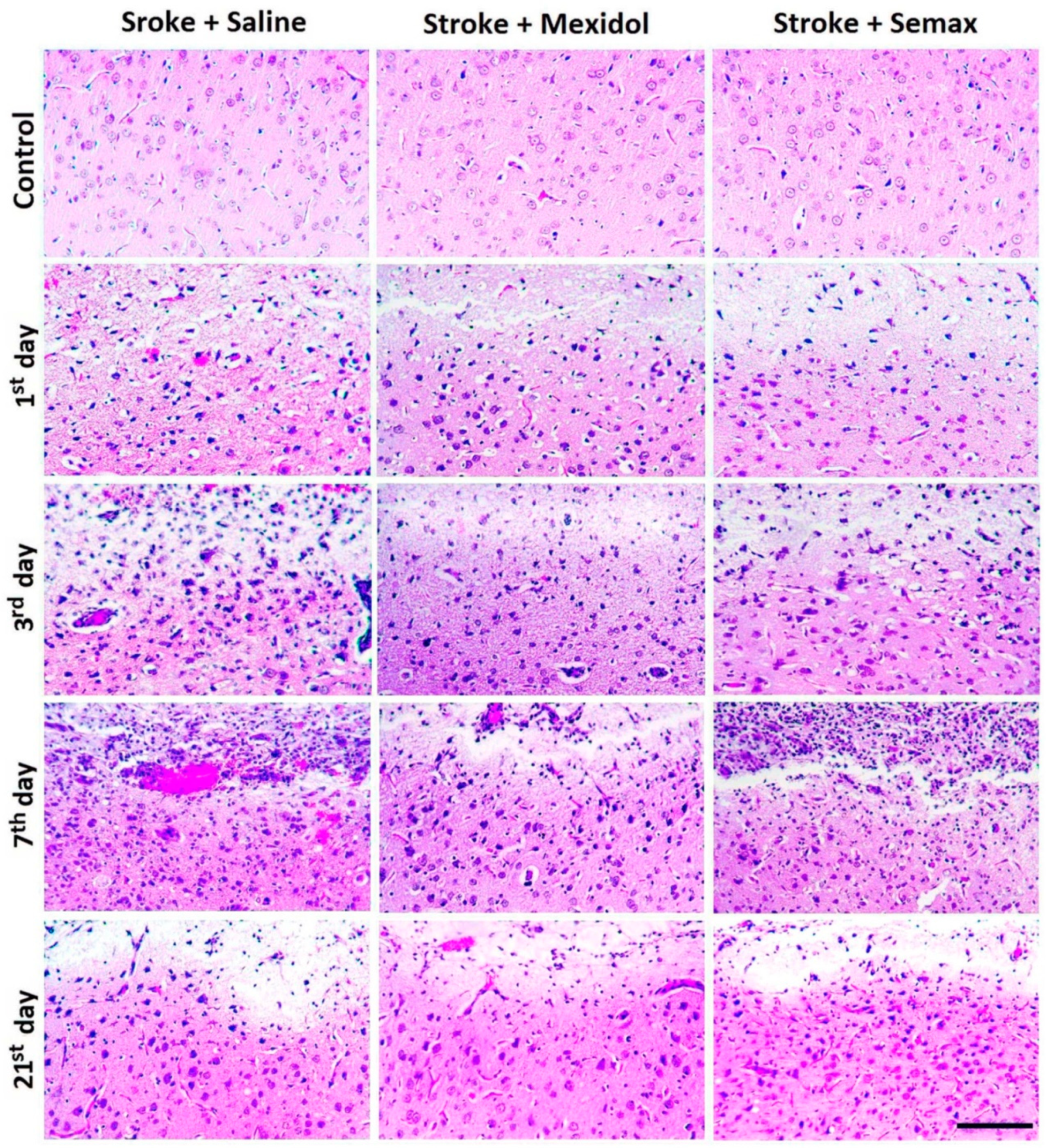
3.1. The Influence of Mexidol and Semax Treatment on the Brain Injury and the Size and the Cellular Composition of the Penumbra
3.2. The Influence of Mexidol and Semax Treatment on the Expression and Activity of PGC-1α in the Penumbral Zone
3.3. Effects of Mexidol and Semax Courses on the Expression and Nuclear Translocation of PGC-1α in the Neurons of the Penumbral Zone
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| AMP | adenosine monophosphate |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AMPK | AMP-activated protein kinase |
| ATP5A | ATP sythase (F1-ATPase) alpha subunit |
| BBB | blood–brain barrier |
| Bcl-2 | B-cell lymphoma 2 anti-apoptotic protein |
| BDNF | brain-derived neurotrophic factor |
| CaMK | Ca2+/calmodulin-dependent protein kinase |
| cAMP | cyclic adenosine monophosphate |
| CAT | catalase |
| CNS | central nervous system |
| COX2 | cytochrome c oxidase subunit 2 |
| CRE | cyclic AMP response element |
| CREB | cyclic AMP response element binding protein |
| cyt c1 | cytochrome c1 |
| DAMP | damage-associated molecular patterns |
| ERR | estrogen-related receptor |
| GABAR | gamma-aminobutyric acid receptor |
| GHs | glucocorticoid hormones |
| GPR 91 | G-protein-coupled receptor |
| GPXs | glutathione peroxidases |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| IL-10 | interleukin-10 |
| IR | immunoreactivity |
| MAPK | mitogen-activated protein kinase |
| MCR | melanocortin receptor |
| Mfn | mitofusin |
| MYC | cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain |
| NAD | nicotinamide adenine dinucleotide |
| NDUFV2 | NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial |
| NMDAR | N-methyl-D-aspartate receptor |
| NRF | nuclear respiratory factor |
| PBS | phosphate buffered saline |
| PC | prefrontal cortex |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PI | photothrombotic ischemia |
| PI3K | phosphoinositide 3-kinases |
| PPAR | peroxisome proliferator-activated receptor |
| SDHA | succinate dehydrogenase, subunit A |
| SIRT1 | silent information regulator two proteins (sirtuins) |
| SOD | superoxide dismutase |
| SP1 | specificity protein 1 |
| SUCNR1 | succinate receptor 1 |
| SYP | synaptophysin |
| TFAM | mitochondrial transcription factor A |
| TGF-β | transforming growth factor beta |
| THs | thyroid hormones |
| TRs | thyroid hormone receptors |
| VEC | vascular endothelial cadherin |
| VEGF | vascular endothelial growth factor |
References
- Avan, A.; Digaleh, H.; Napoli, M.D.; Stranges, S.; Behrouz, R.; Shojaeianbabaei, G.; Amiri, A.; Tabrizi, R.; Mokhber, N.; Spence, J.D.; et al. Socioeconomic status and stroke incidence, prevalence, mortality, and worldwide burden: An ecological analysis from the Global Burden of Disease Study 2017. BMC Med. 2019, 17, 191–221. [Google Scholar] [CrossRef]
- Handschin, C. The biology of PGC-1 alpha and its therapeutic potential. Trends Pharm. Sci. 2009, 30, 322–329. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Puddifoot, C.; Martel, M.-A.; Soriano, F.X.; Camacho, A.; Vidal-Puig, A.; Wyllie, D.J.A.; Hardingham, G.E. PGC-1α negatively regulates extrasynaptic NMDAR activity and excitotoxicity. J. Neurosci. 2012, 32, 6995–7000. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2014, 282, 647–672. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1 α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A. Mitofusins 1/2 and ERR alpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as nuclear receptors for nutrient and energy metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Li, X.; Lindsley, L.; Wang, X.; Shaughnessy, S.; Daniels, T.G.; Szustakowski, J.; Nirmala, N.R.; Wu, Z.; Stevenson, S.C. Estrogen-related receptor alpha is essential for the expression of antioxidant protection genes and mitochondrial function. Biochem. Biophys. Res. Commun. 2007, 357, 231–236. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Villena, J.A.; Kralli, A. ERRα: A metabolic function for the oldest orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef]
- Cowell, R.; Blake, K.R.; Russel, J.W. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J. Comp. Neurol. 2007, 502, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A.; Mastaitis, J.W.; Kokkotou, E.G.; Puigserver, P.; Spiegelman, B.M.; Maratos-Flier, E. Characterization of the peroxisome proliferator activated receptor coactivator 1 alpha (PGC 1alpha) expression in the murine brain. Brain Res. 2003, 961, 255–260. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250–1266. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. Mitochondria and PGC-1α in aging and age-associated diseases. J. Aging Res. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Wong-Riley, M.T. Coupling of energy metabolism and synaptic transmission at the transcriptional level: Role of nuclear respiratory factor 1 I regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 2009, 29, 483–492. [Google Scholar] [CrossRef]
- Li, Z.; Cogswell, M.; Hixson, K.; Brooks-Kayal, A.R.; Russek, S.J. Nuclear Respiratory Factor 1 (NRF-1) controls the activity dependent transcription of the GABA-A receptor beta 1 subunit gene in neurons. Front. Mol. Neurosci. 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Dason, E.S.; Zanon-Moreno, V.; Jiang, Q.; Nahirnyj, A.; Chan, D.; Flanagan, J.G.; Sivak, J.M. PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am. J. Pathol. 2014, 184, 1017–1029. [Google Scholar] [CrossRef]
- Eisele, P.S.; Salatino, S.; Sobek, J.; Hottiger, M.O.; Handschin, C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 2013, 288, 2246–2260. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox. Sign. 2010, 13, 1011–1022. [Google Scholar] [CrossRef]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; van der Pol, S.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170–183. [Google Scholar] [CrossRef]
- Shulga, A.; Blaesse, A.; Kysenius, K.; Huttunen, H.J.; Tanhuanpää, K.; Saarma, M. Thyroxin regulates BDNF expression to promote survival of injured neurons. Mol. Cell Neurosci. 2009, 42, 408–418. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1 beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007, 274, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Tokudome, S.; Shimizu, N.; Yoshikawa, N.; Ogawa, C.; Shirakawa, K. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor c coactivator 1a. J. Biol. Chem. 2007, 282, 25970–25980. [Google Scholar] [CrossRef]
- Petegnief, V.; Planas, A.M. SIRT1 regulation modulates stroke outcome. Transl. Stroke Res. 2013, 4, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Consoli, A.; Formoso, G. Do thiazolidinediones still have a role in treatment of type 2 diabetes mellitus? Diabetes Obes. Metab. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhäusl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef]
- Selvin, E.; Bolen, S.; Yeh, H.C.; Wiley, C.; Wilson, L.M.; Marinopoulos, S.S. Cardiovascular outcomes in trials of oral diabetes medications: A systematic review. Arch. Intern. Med. 2008, 168, 2070–2080. [Google Scholar] [CrossRef]
- Ohira, M.; Endo, K.; Saiki, A.; Miyashita, Y.; Terai, K.; Murano, T. Atorvastatin and pitavastatin enhance lipoprotein lipase production in L6 skeletal muscle cells through activation of adenosine monophosphate-activated protein kinase. Metabolism 2012, 61, 1452–1460. [Google Scholar] [CrossRef]
- Guan, X.; Wang, Y.; Kai, G.; Zhao, S.; Huang, T.; Li, Y.; Xu, Y.; Zhang, L.; Pang, T. Cerebrolysin ameliorates focal cerebral ischemia injury through neuroinflammatory inhibition via CREB/PGC-1α pathway. Front. Pharmacol. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Povarnina, P.; Logvinov, I.O.; Antipova, T.A.; Seredenin, S.B. Mimetics of brain-derived neurotrophic factor loops 1 and 4 are active in a model of ischemic stroke in rats. Drug Des. Devel. Ther. 2016, 10, 3545–3553. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Povarnina, P.Y.; Volkova, A.A.; Kruglov, S.V.; Antipova, T.A.; Seredenin, S.B. A nerve growth factor dipeptide mimetic stimulates neurogenesis and synaptogenesis in the hippocampus and striatum of adult rats with focal cerebral ischemia. Acta Nat. 2019, 11, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.I.; Xiong, L.-J.; Tong, Y.U.; Mao, M. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed. Rep. 2013, 1, 167–176. [Google Scholar] [CrossRef]
- Martynov, M.Y.; Kostenko, E.V.; Petrova, L.V.; Bobyreva, S.N. The efficacy of Semax in the tretament of patients at different stages of ischemic stroke. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2018, 118, 61–68. (In Russian) [Google Scholar] [CrossRef]
- Gusev, E.I.; Skvortsova, V.I.; Chukanova, E.I. Semax in prevention of disease progress and development of exacerbations in patients with cerebrovascular insufficiency. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2005, 105, 35–40. (In Russian) [Google Scholar] [PubMed]
- Romanova, G.A.; Silachev, D.N.; Shakova, F.M.; Kvashennikova, Y.N.; Viktorov, I.V.; Shram, S.I.; Myasoedov, N.F. Neuroprotective and antiamnesic effects of Semax during experimental ischemic infarction of the cerebral cortex. Bull. Exp. Biol Med. 2006, 142, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Agapova, T.I.; Agniullin, I.V.; Silachev, D.N.; Shadrina, M.I.; Slominskii, P.A.; Shram, I.; Limborskaia, S.A.; Miasoedov, N.F. Effect of Semax on the temporary dynamics of brain-derived neurotrophic factor and nerve growth factor gene expression in the rat hippocampus and frontal cortex. Mol. Gen. Mikrobiol. Virusol. 2008, 3, 28–32. [Google Scholar]
- Dmitrieva, V.G.; Povarinova, V.I.; Limborska, S.A.; Myasoedov, N.F.; Dergunova, L.V. Semax and pro-gly-pro activate the transcription of neurotrophins and their receptor genes after cerebral ischemia. Cell Mol. Neurobiol. 2010, 30, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Sudarkina, O.Y.; Dmitrieva, V.G.; Gubsky, L.V. Novel insights into the protective properties of ACTH(4-7)PGP (Semax) peptide at the transcriptome level following cerebral ischaemia–reperfusion in rats. Genes 2020, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, E.V.; Dmitrieva, V.G.; Povarova, O.V.; Limborska, S.A.; Skvortsova, V.I.; Myasoedov, N.F.; Dergunova, L.V. The peptide Semax affects the expression of genes related to the immune and vascular systems in rat brain focal ischemia: Genome-wide transcriptional analysis. BMC Genom. 2014, 15, 228–240. [Google Scholar] [CrossRef]
- Medvedeva, E.V.; Dmitrieva, V.G.; Limborska, S.A.; Myasoedov, N.F.; Dergunova, L.V. Semax, an analog of ACTH(4-7), regulates expression of immune response genes during ischemic brain injury in rats. Mol. Genet. Genom. 2017, 292, 635–653. [Google Scholar] [CrossRef]
- Gallo-Payet, N. Adrenal and extra-adrenal functions of ACTH. J. Mol. Endocrinol. 2016, 56, T135–T156. [Google Scholar] [CrossRef] [PubMed]
- Lisak, R.P.; Benjamins, J. Melanocortins, melanocortin receptors and multiple sclerosis. Brain Sci. 2017, 7, 104. [Google Scholar] [CrossRef]
- Lyang, O.V.; Kochetov, A.G. The use of Mexidol in chronic cerebral ischemia. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2013, 113, 126–129. (In Russian) [Google Scholar]
- Kutashov, V.A.; Ulyanova, O.V. The study of the efficacy and safety of Mexidol and Mexidol Forte 250 in patients with chronic cerebral ischemia. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2019, 119, 89–92. (In Russian) [Google Scholar] [CrossRef] [PubMed]
- Androfagina, O.V.; Kuznetsova, T.V.; Svetkina, A.A. Mexidol in the rehabilitation of patients in the acute ischemic stroke. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2015, 115, 77–79. (In Russian) [Google Scholar] [CrossRef] [PubMed]
- Stakhovskaya, L.V.; Tyutyumova, E.A.; Fedin, A.I. Modern approaches to neuroprotective treatment of ischemic stroke. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2017, 8, 75–80. (In Russian) [Google Scholar] [CrossRef]
- Chekman, I.S.; Belenichev, I.F.; Yakovleva, I.Y.; Gorchakova, N.A.; Buhtiyarova, N.V.; Morguntsova, S.A.; Brazhko, O.O.; Levich, S.V. Influence of Mexidol on early genomic response and morphofunctional parameters of the brain cortex sensorimotor zone neurons after arteria carotis communis occlusion. Oxid. Antioxid. Med. Sci. 2015, 4, 33–38. [Google Scholar] [CrossRef]
- Voronina, T.A. Mexidol: The spectrum of pharmacological effects. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2012, 12, 86–90. (In Russian) [Google Scholar]
- Hamel, D.; Sanchez, M.; Duhamel, F.; Roy, O.; Honore, J.-C.; Noueihed, B.; Zhou, T.; Nadeau-Vallee, M.; Hou, X.; Lavoie, J.C.; et al. G-protein–coupled receptor 91 and succinate are key contributors in neonatal postcerebral hypoxia-ischemia recovery. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 285–293. [Google Scholar] [CrossRef]
- Sapieha, P.; Sirinyan, M.; Hamel, D.; Zaniolo, K.; Joyal, J.-S.; Cho, J.-H.; Honoré, J.-C.; Kermorvant-Duchemin, E.; Varma, D.R.; Tremblay, S. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 2008, 14, 1067–1076. [Google Scholar] [CrossRef]
- Kirova, Y.I.; Shakova, F.M.; Germanova, E.L.; Romanova, G.A.; Voronina, T.A. The effect of Mexidol on cerebral mitochondriogenesis at a young age and during aging. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova [S.S. Korsakov J. Neurol. Psychiatry] 2020, 120, 62–69. (In Russian) [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: San Diego, CA, USA, 1998; p. 474. [Google Scholar]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Morini, M.F.; Giampietro, C.; Corada, M.; Pisati, F.; Lavarone, E.; Cunha, S.I. VE-Cadherin–mediated epigenetic regulationof endothelial gene expression. Circ. Res. 2018, 122, 231–245. [Google Scholar] [CrossRef]
- Vestweber, D. VE-Cadherin the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arter. Thromb. Vasc. Biol 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Bras, A.L.; Lionneton, F.; Mattot, V.; Lelievre, E.; Caetano, B.; Spruyt, N.; Soncin, F. HIF-2a specifically activates the VE-cadherin promoter independently of hypoxia and in synergy with Ets-1 through two essential ETS-binding sites. Oncogene 2007, 26, 7480–7489. [Google Scholar] [CrossRef]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Shoag, J.; Arany, Z. Regulation of hypoxia-inducible genes by PGC-1α. Arter. Thromb. Vasc. Biol. 2010, 30, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.-N.; Zhu, H.; Zhang, H.-J.; Zhang, W.-F.; Jin, H.-L.; Wang, L. HIF-1α induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J. Gastroenterol. 2014, 20, 17894–17904. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1a and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Shram, S.I.; Shakova, F.M.; Romanova, G.A.; Miasoedov, N.F. Formation of the spatial memory in rats with ischemic injury in prefrontal areas of the cortex-effects of a sinthetic analogue of ACTH(4-7). Zh. Vyss. Nerv. Deiat. Im. I. P. Pavlova [I.P. Pavlov. J. High. Nerv. Act. ] 2008, 58, 458–466. (In Russian) [Google Scholar]
- Du, J.; Ma, M.; Zhao, Q.; Fang, L.; Chang, J.; Wang, Y. Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience 2013, 231, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Jiang, W.; Liu, H.; Wang, J.; Zheng, K.; Cui, P.; Feng, Y.; Dang, C.; Bu, Y.; Wang, Q.M.; et al. Upregulation of neuronal PGC-1α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 2020, 10, 2832–2848. [Google Scholar] [CrossRef] [PubMed]
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Shi, H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med. Chem. 2009, 16, 4593–4600. [Google Scholar] [CrossRef] [PubMed]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225–a014241. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef]
- Povarnina, P.Y.; Volkova, A.A.; Gudasheva, T.A.; Seredenin, S.B. Comparison of the pharmacological effects of dimeric dipeptide nerve growth factor mimetic gk-2 and Mexidol on the model of ischemic stroke in rats. Bull. Exp. Biol. Med. 2017, 164, 173–176. [Google Scholar] [CrossRef]
- Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L. SUCNR1-mediated chemotaxis of macrophages aggravates obesi-ty-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2015, 53, 1181–1194. [Google Scholar] [CrossRef]
- Grigorjeva, M.E.; Lyapina, L.A. Anticoagulation and antiplatelet effects of Semax under conditions of acute and chronic immobilization stress. Bull. Exp. Biol. Med. 2010, 149, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Vaccari, J.P.R.; Dietrich, W.D.; Popovich, P.G.; Keanec, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Julier, Z.; Park, A.J.; Briquez, P.S.; Martino, M.M. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017, 53, 13–28. [Google Scholar] [CrossRef]
- Igarashi, J.; Okamoto, R.; Yamashita, T.; Hashimoto, T.; Karita, S.; Kozo, N.; Kubota, Y.; Takata, M.; Yamaguchi, F.; Tokuda, M.; et al. A key role of PGC-1a transcriptional coactivator in production of VEGF by a novel angiogenic agent COA-Cl in cultured human fibroblasts. Physiol. Rep. 2016, 4, e12742–e12752. [Google Scholar] [CrossRef] [PubMed]
- Ariza, A.C.; Deen, P.M.T.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012, 3, 1–8. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.P.; Lin, D.C.H.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G- protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and rennin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar]
- Lukyanova, L.D.; Kirova, Y.I. Mitochondria-controlled signalling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 1–13. [Google Scholar] [CrossRef]
- Diskin, C.; Palsson-McDermott, E.M. Metabolic modulation in macrophage effector function. Front. Immunol. 2018, 9, 270–287. [Google Scholar] [CrossRef]
- De la Vega, M.C.; Burda, J.; Nemethova, M.; Quevedo, C.; Alcázar, A.; Martín, M.E. Possible mechanisms involved in the down-regulation of translation during transient global ischaemia in the rat brain. Biochem. J. 2001, 357, 819–826. [Google Scholar] [CrossRef]
- Khunderyakova, N.V.; Zacharchenko, M.V.; Zakharchenko, A.V.; Kondrashova, M.N. Hyperactivation of succinate dehydrogenase in lymphocytes of newborn rats. Biochemistry 2008, 73, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef]
- Garman, R.H. Histology of the central nervous system. Toxicol. Pathol. 2011, 39, 22–35. [Google Scholar] [CrossRef]
- Parker, G.A.; Picut, C.A. Atlas of Histology of the Juvenile Rats, 1st ed.; Elsevier: London, UK, 2016; p. 462. [Google Scholar]
- Hasegawa, A.; Furuhama, K. Atlas of the Hematology of the Laboratory Rat, 1st ed.; Elsevier Science Ltd.: London, UK, 1998; p. 171. [Google Scholar]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef]
- Haniu, H.; Watanabe, D.; Kawashima, Y.; Matsumoto, H. Two-dimensional gel-based protein standardization verified by western blot analysis. In Western Blotting. Methods and Protocols; Kurien, B.T., Scofield, R.H., Eds.; Springer Science + Business Media: New York, NY, USA, 2015; pp. 473–480. [Google Scholar]
- Li, R.; Shen, Y. An old method facing a new challenge: Re-visiting housekeeping proteins as internal reference control for neuroscience research. Life Sci. 2013, 92, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [Google Scholar] [PubMed]
- Lind, D.; Franken, S.; Kappler, J.; Jankowski, J.; Schilling, K. Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J. Neurosci Res. 2005, 79, 295–302. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakova, F.M.; Kirova, Y.I.; Silachev, D.N.; Romanova, G.A.; Morozov, S.G. Protective Effects of PGC-1α Activators on Ischemic Stroke in a Rat Model of Photochemically Induced Thrombosis. Brain Sci. 2021, 11, 325. https://doi.org/10.3390/brainsci11030325
Shakova FM, Kirova YI, Silachev DN, Romanova GA, Morozov SG. Protective Effects of PGC-1α Activators on Ischemic Stroke in a Rat Model of Photochemically Induced Thrombosis. Brain Sciences. 2021; 11(3):325. https://doi.org/10.3390/brainsci11030325
Chicago/Turabian StyleShakova, Fatima M., Yuliya I. Kirova, Denis N. Silachev, Galina A. Romanova, and Sergey G. Morozov. 2021. "Protective Effects of PGC-1α Activators on Ischemic Stroke in a Rat Model of Photochemically Induced Thrombosis" Brain Sciences 11, no. 3: 325. https://doi.org/10.3390/brainsci11030325
APA StyleShakova, F. M., Kirova, Y. I., Silachev, D. N., Romanova, G. A., & Morozov, S. G. (2021). Protective Effects of PGC-1α Activators on Ischemic Stroke in a Rat Model of Photochemically Induced Thrombosis. Brain Sciences, 11(3), 325. https://doi.org/10.3390/brainsci11030325