
Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience

 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
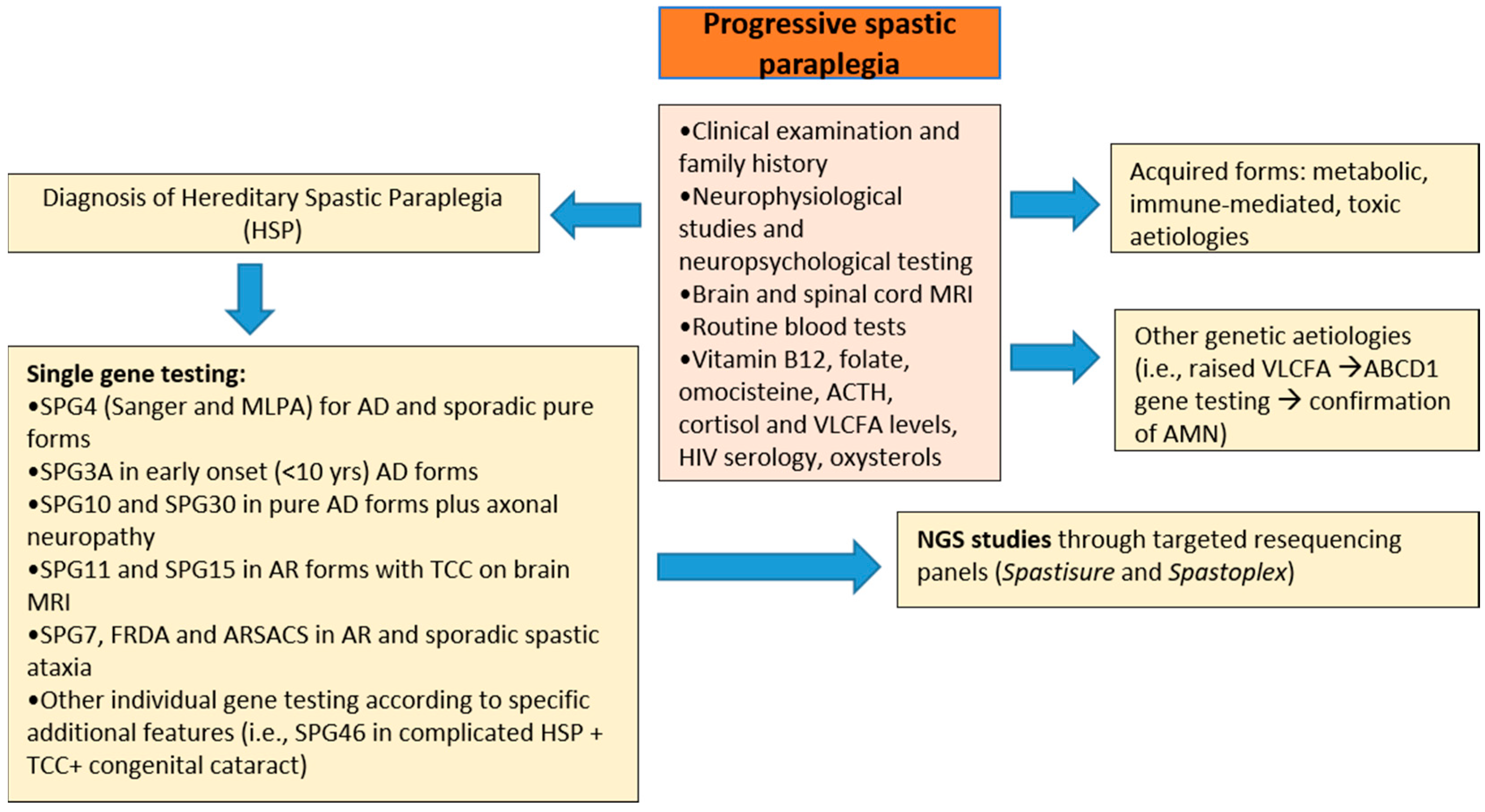
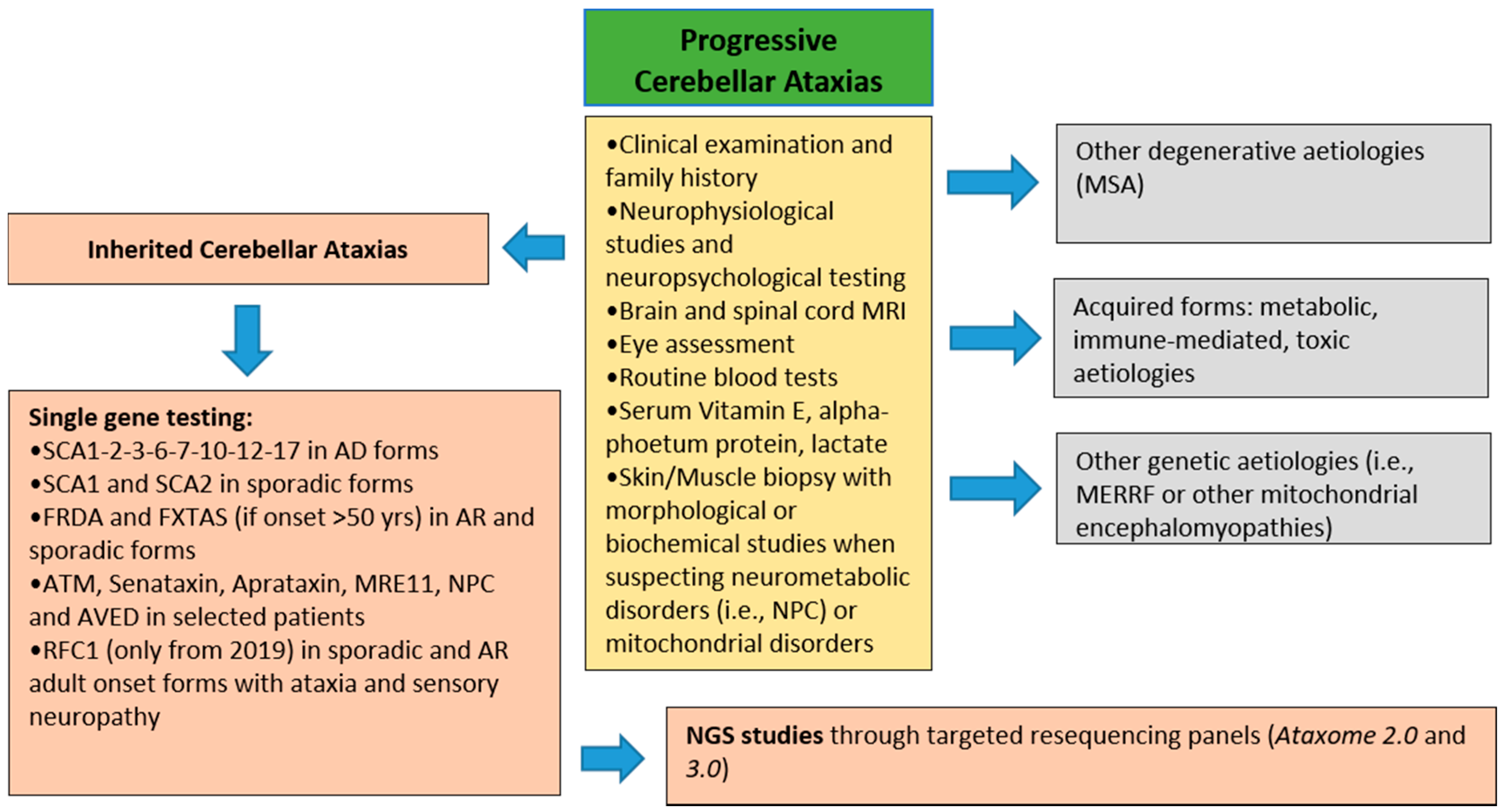
2. Materials and Methods
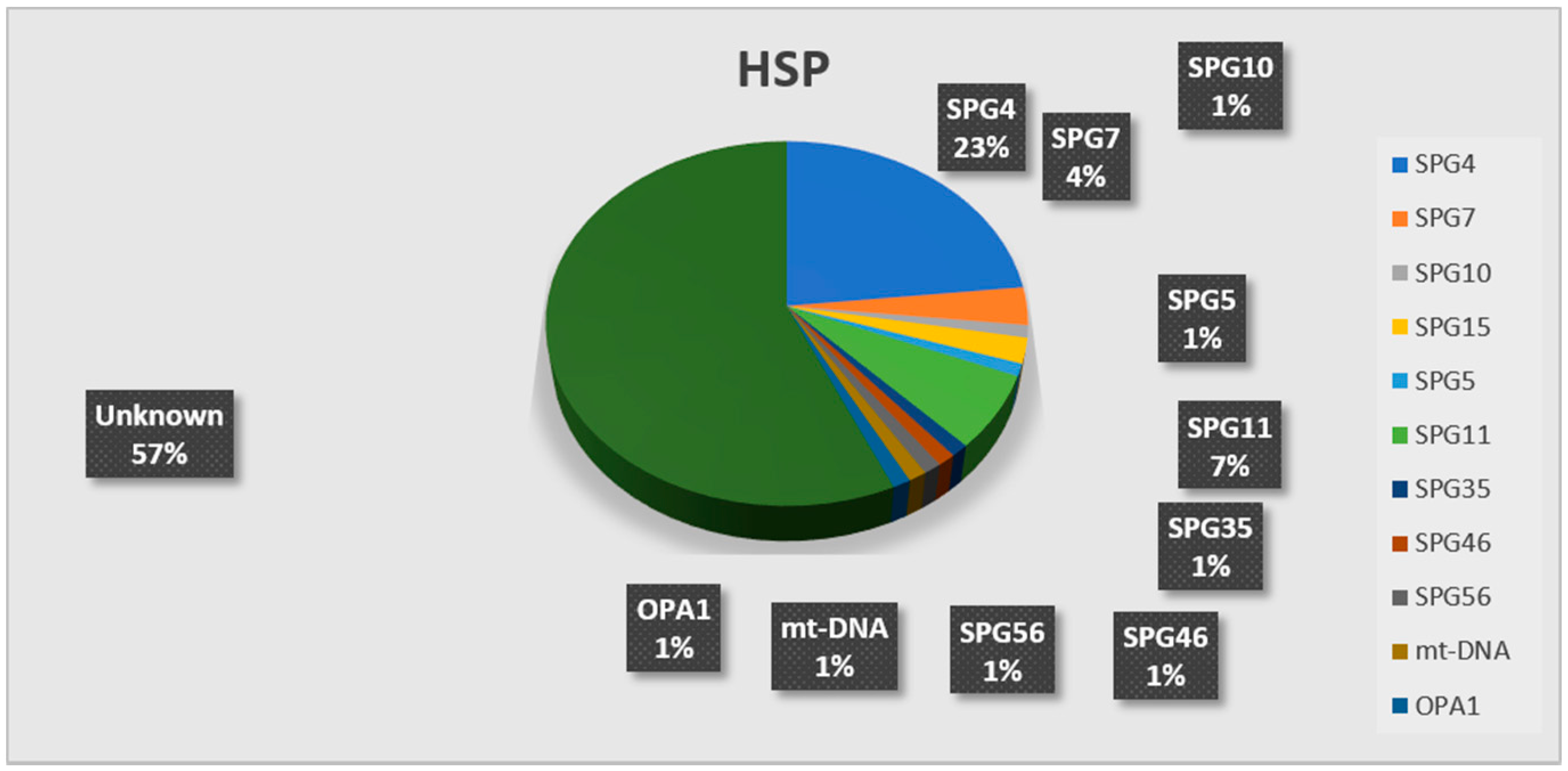
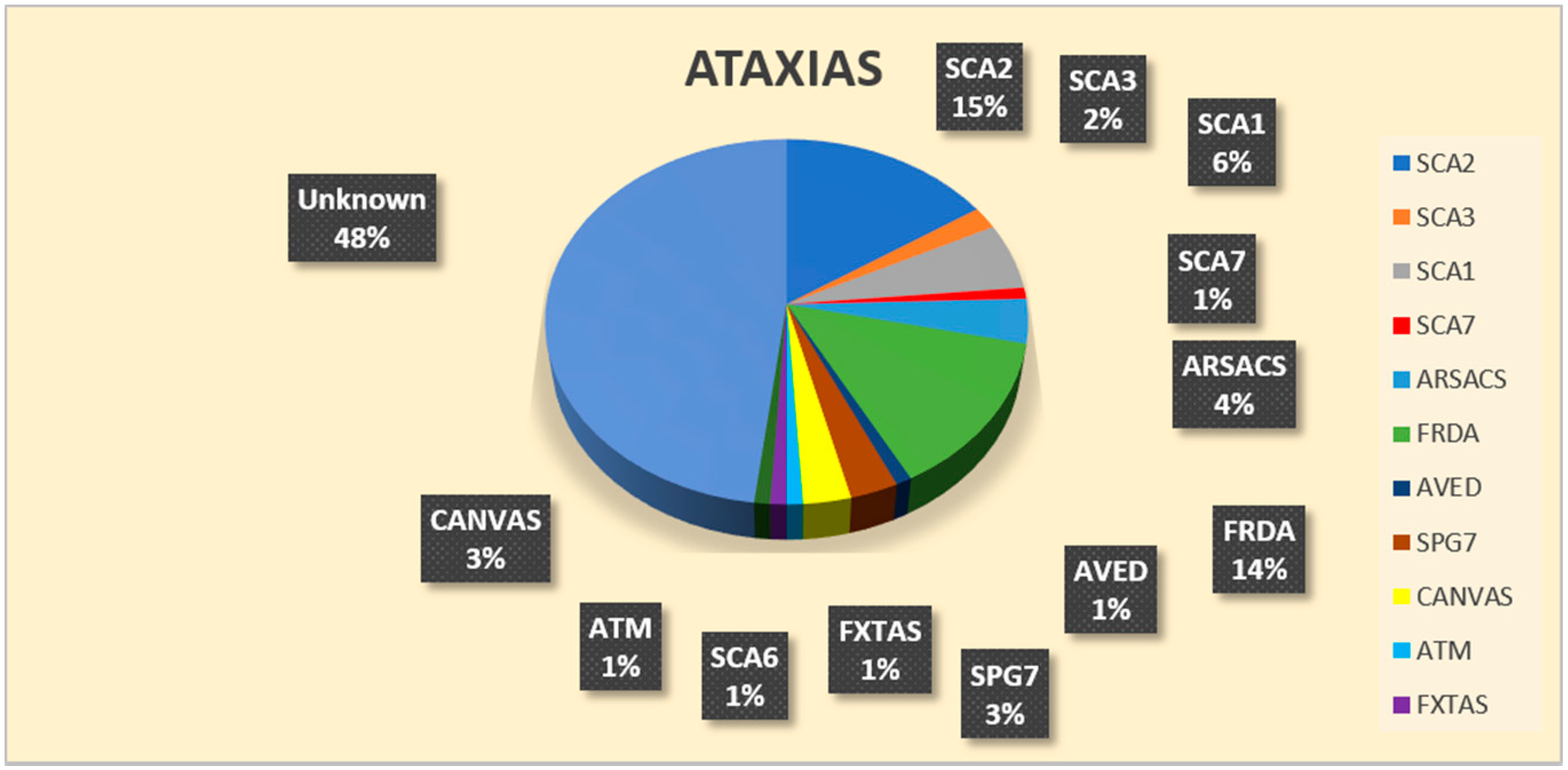
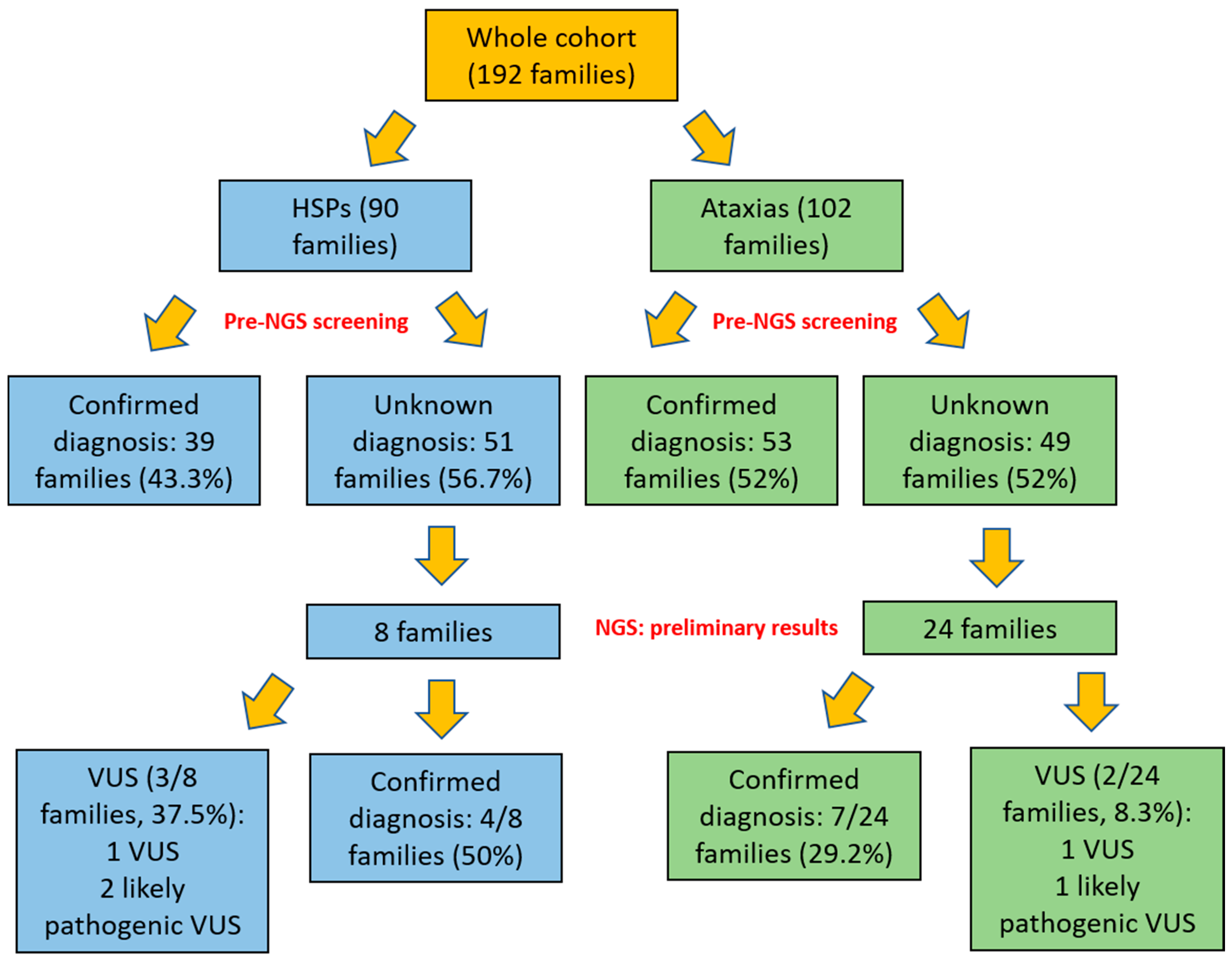
3. Results
NGS Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Hensiek, A.; Kirker, S.; Reid, E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J. Neurol. 2015, 262, 1601–1612. [Google Scholar] [CrossRef]
- Parodi, L.; Coarelli, G.; Stevanin, G.; Brice, A.; Durr, A. Hereditary ataxias and paraparesias: Clinical and genetic update. Curr. Opin. Neurol. 2018, 31, 462–471. [Google Scholar] [CrossRef]
- Racis, L.; Storti, E.; Pugliatti, M.; Agnetti, V.; Tessa, A.; Santorelli, F.M. Novel SPAST deletion and reduced DPY30 expression in a spastic paraplegia type 4 kindred. BMC Med Genet. 2014, 15, 39. [Google Scholar] [CrossRef]
- Burguez, D.; Polese-Bonatto, M.; Scudeiro, L.A.J.; Björkhem, I.; Schöls, L.; Jardim, L.B.; Matte, U.; Saraiva-Pereira, M.L.; Siebert, M.; Saute, J.A.M. Clinical and molecular characterization of hereditary spastic paraplegias: A next-generation sequencing panel approach. J. Neurol. Sci. 2017, 383, 18–25. [Google Scholar] [CrossRef]
- Dong, E.-L.; Wang, C.; Wu, S.; Lu, Y.-Q.; Lin, X.-H.; Su, H.-Z.; Zhao, M.; He, J.; Ma, L.-X.; Wang, N.; et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chrestian, N.; Dupré, N.; Gan-Or, Z.; Szuto, A.; Chen, S.; Venkitachalam, A.; Brisson, J.-D.; Warman-Chardon, J.; Ahmed, S.; Ashtiani, S.; et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2017, 3, e122. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; SPATAX Consortium; Rosa, A.L.; Lerer, I.; Hamri, A.; et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2007, 131, 772–784. [Google Scholar] [CrossRef]
- Esteves, T.; Durr, A.; Mundwiller, E.; Loureiro, J.L.; Boutry, M.; Gonzalez, M.A.; Gauthier, J.; El-Hachimi, K.H.; Depienne, C.; Muriel, M.-P.; et al. Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am. J. Hum. Genet. 2014, 94, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Goizet, C.; Durr, A.; Habarou, F.; Morais, S.; Dionne-Laporte, A.; Tao, F.; Konop, J.; Stoll, M.; Charles, P.; et al. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain 2015, 138, 2191–2205. [Google Scholar] [CrossRef]
- Erlich, Y.; Edvardson, S.; Hodges, E.; Zenvirt, S.; Thekkat, P.; Shaag, A.; Dor, T.; Hannon, G.J.; Elpeleg, O. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011, 21, 658–664. [Google Scholar] [CrossRef]
- Lee, J.R.; Srour, M.; Kim, D.; Hamdan, F.F.; Lim, S.H.; Brunel-Guitton, C.; Décarie, J.C.; Rossignol, E.; Mitchell, G.A.; Schreiber, A.; et al. De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum. Mutat. 2015, 36, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ferrero, E.; Coto, E.; Beetz, C.; Gamez, J.; Corao, A.; Diaz, M.; Esteban, J.; Del Castillo, E.; Moris, G.; Infante, J.; et al. SPG7mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V. Clin. Genet. 2013, 83, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. SPG7 Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135 Pt 10, 2980–2993. [Google Scholar] [CrossRef]
- Rydning, S.L.; Dudesek, A.; Rimmele, F.; Funke, C.; Krüger, S.; Biskup, S.; Vigeland, M.D.; Hjorthaug, H.S.; Sejersted, Y.; Tallaksen, C.; et al. A novel heterozygous variant inERLIN2causes autosomal dominant pure hereditary spastic paraplegia. Eur. J. Neurol. 2018, 25, 943-e71. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef]
- Beaudin, M.; Klein, C.J.; Rouleau, G.A.; Dupré, N. Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias 2017, 4, 1–12. [Google Scholar] [CrossRef]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Shribman, S.; Reid, E.; Crosby, A.H.; Houlden, H.; Warner, T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019, 18, 1136–1146. [Google Scholar] [CrossRef]
- Galatolo, D.; Tessa, A.; Filla, A.; Santorelli, F.M. Clinical application of next generation sequencing in hereditary spinocerebellar ataxia: Increasing the diagnostic yield and broadening the ataxia-spasticity spectrum. A retrospective analysis. Neurogenetics 2018, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Schüle, R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Lee, H.; Deignan, J.L.; Strom, S.P.; Kantarci, S.; Wang, X.; Quintero-Rivera, F.; Vilain, E.; Grody, W.W.; Perlman, S.; et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014, 71, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, A.; Tessa, A.; Casali, C.; Dotti, M.T.; Filla, A.; Silvestri, G.; Antenora, A.; Astrea, G.; Barghigiani, M.; Battini, R.; et al. Next generation molecular diagnosis of hereditary spastic paraplegias: An Italian cross-sectional study. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tonduti, D.; Aiello, C.; Renaldo, F.; Dorboz, I.; Saaman, S.; Rodriguez, D.; Fettah, H.; Elmaleh-Bergès, M.; Biancheri, R.; Barresi, S.; et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur. J. Paediatr. Neurol. 2016, 20, 323–330. [Google Scholar] [CrossRef]
- Riso, V.; Rossi, S.; Perna, A.; Nicoletti, T.; Bosco, L.; Zanni, G.; Silvestri, G. NGS-based detection of a novel mutation in PRKCG (SCA14) in sporadic adult-onset ataxia plus dystonic tremor. Neurol. Sci. 2020, 41, 2989–2991. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Tessa, A.; Silvestri, G.; De Leva, M.F.; Modoni, A.; Denora, P.S.; Masciullo, M.; Dotti, M.T.; Casali, C.; Melone, M.A.B.; Federico, A.; et al. A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. J. Neurol. 2008, 255, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Masciullo, M.; Tessa, A.; Perazza, S.; Santorelli, F.; Perna, A.; Silvestri, G. Hereditary spastic paraplegia: Novel mutations and expansion of the phenotype variability in SPG56. Eur. J. Paediatr. Neurol. 2016, 20, 444–448. [Google Scholar] [CrossRef]
- Lieto, M.; Riso, V.; Galatolo, D.; De Michele, G.; Rossi, S.; Barghigiani, M.; Cocozza, S.; Pontillo, G.; Trovato, R.; Saccà, F.; et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur. J. Neurol. 2020, 27, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Nahhas, N.; Conant, A.; Hamilton, E.; Curiel, J.; Simons, C.; van der Knaap, M.; Vanderver, A. TUBB4A-Related Leukodystrophy. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016; 1993–2020; PMID: 27809427. [Google Scholar]
- Delplanque, J.; Devos, D.; Huin, V.; Genet, A.; Sand, O.; Moreau, C.; Goizet, C.; Charles, P.; Anheim, M.; Monin, M.L.; et al. TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain 2014, 137, 2657–2663. [Google Scholar] [CrossRef]
- Zhao, C.; Takita, J.; Tanaka, Y.; Setou, M.; Nakagawa, T.; Takeda, S.; Yang, H.W.; Terada, S.; Nakata, T.; Takei, Y.; et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1B-beta. Cell 2001, 105, 587–597, Erratum in 2001, 106, 127. [Google Scholar] [CrossRef]
- Perna, A.; Masciullo, M.; Modoni, A.; Cellini, E.; Parrini, E.; Ricci, E.; Donati, A.M.; Silvestri, G. Severe 5,10-methylenetetrahydrofolate reductase deficiency: A rare, treatable cause of complicated hereditary spastic paraplegia. Eur. J. Neurol. 2018, 25, 602–605. [Google Scholar] [CrossRef]
- Németh, A.H.; Kwasniewska, A.C.; Lise, S.; Schnekenberg, R.P.; Becker, E.B.E.; Bera, K.D.; Shanks, M.E.; Gregory, L.; Buck, D.; Cader, M.Z.; et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013, 136, 3106–3118. [Google Scholar] [CrossRef]
- Elert-Dobkowska, E.; Stepniak, I.; Krysa, W.; Ziora-Jakutowicz, K.; Rakowicz, M.; Sobanska, A.; Pilch, J.; Antczak-Marach, D.; Zaremba, J.; Sulek, A. Next-generation sequencing study reveals the broader variant spectrum of hereditary spastic paraplegia and related phenotypes. Neurogenetics 2019, 20, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.-L.; Davoine, C.-S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Lu, C.; Li, L.-X.; Dong, H.-L.; Wei, Q.; Liu, Z.-J.; Ni, W.; Gitler, A.D.; Wu, Z.-Y. Targeted next-generation sequencing improves diagnosis of hereditary spastic paraplegia in Chinese patients. J. Mol. Med. 2018, 96, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.S.; Koutsis, G.; Tucci, A.; Panas, M.; Baklou, M.; Breza, M.; Karadima, G.; Houlden, H. Hereditary spastic paraplegia in Greece: Characterisation of a previously unexplored population using next-generation sequencing. Eur. J. Hum. Genet. 2015, 24, 857–863. [Google Scholar] [CrossRef]
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016, 139 Pt 7, 1904–1918. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, Y.-J.; Wang, M.-W.; Lin, X.-H.; Dong, E.-L.; Chen, W.-J.; Wang, N.; Lin, X. Genetic and clinical profile of chinese patients with autosomal dominant spastic paraplegia. Mol. Diagn. Ther. 2019, 23, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, H.-L.; Pan, L.-Y.; Chen, C.-X.; Yan, Y.-T.; Wang, R.-M.; Li, H.-F.; Liu, Z.-J.; Tao, Q.-Q.; Wu, Z.-Y. Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl. Neurodegener. 2019, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSP (90 Families) | |||
| AD 30/90 (33.3%) | AR 27/90 (30%) | Sporadic 33/90 (36.7%) | |
| diagnosis reached | 17/30 (56.7%) | 15/27 (55.5%) | 7/33 (21.2%) |
| genetic diagnosis | 16 SPG4/30 SPG4 SGT 1 SPG10/1 SPG10 SGT | 6 SPG11/8 SPG11 SGT 2 SPG15/2 SPG15 SGT 2 SPG7/5 SPG7 SGT 1 SPG5/1 SPG5 SGT 1 SPG35/1 SPG35 SGT 1 SPG45/1 SPG45 SGT 1 SPG56/4 SPG56 SGT 1 MT-ND3/1 mtDNA sequencing | 5 SPG4/33 SPG4 SGT 1 SPG7/6 SPG7 SGT 1 OPA1/1 OPA1 SGT |
| CA (102 Families) | |||
| AD 32/102 (31.4%) | AR 31/102 (30.4%) | Sporadic 39/102 (38.2%) | |
| diagnosis reached | 24/32 (75%) | 16/31 (51.6%) | 13/39 (33.3%) |
| genetic diagnosis | 15 SCA2/32 SCA2 SGT 5 SCA1/32 SCA1 SGT 2 SCA3/32 SCA3 SGT 1 SCA6/32 SCA6 SGT 1 SCA7/32 SCA7 SGT | 10 FRDA/31 FRDA SGT 3 SACS/4 SACS SGT 1 ATM/1 ATM SGT 1 AVED/1 TTPA SGT 1 CANVAS/2 RFC1 SGT | 4 FRDA/39 FRDA SGT 3 SPG7/6 SPG7 SGT 2 CANVAS/20 RFC1 SGT 1 SACS/7 SACS SGT 1 SCA1/39 SCA1 SGT 1 SCA2/39 SCA2 SGT 1 FXTAS/6 FMR1 SGT |
| Patient | Age of Onset | Age at Diagnosis | Family History | Phenotype | Neuroimaging | Gene/Disease | Variant |
|---|---|---|---|---|---|---|---|
| #1 [30] | 25 | 25 | AR | Pure HSP + ny | normal | CYP721/SPG5 NM_004820 | c.338insT/p.Phe114fs*3 (Homozygous) |
| #2 [30] | 15 | 37 | AR | HSP, epilepsy and cognitive delay | TCC and spinal cord atrophy | KIAA1840/SPG11 NM_025137 | c.2833A > G/p.Arg945Gly + c.128delC/p.S43fs*15 |
| #3 | 20 | 37 | AR | Pure HSP | normal | PGN/SPG7 NM_003119 | c.1369C > T/p.R457* + c.1617delC/p.V540Cfs*52 |
| #4 | 20 | 35 | AR | HSP + mild hyperCK | Mild TCC | KIAA1840/SPG11 NM_025137 | c.2842-2843insG/p.Val948GlyfsTer6 + c.3291 + 3A > G |
| #5 [30] | 30 | 38 | AR | HSP + dysarthria | Mild CA | POLR3A-related leukodystrophy NM_007055 | c.1909 + 22G > A + c.3201_3202delGC/p.R1069fs*2 |
| #6 | 35 | 57 | AD | Spastic ataxia | CA | PGN/SPG 7 NM_003119.3 | c.1013G > T/p.G338V |
| #7 | 40 | 48 | SP | Progressive spastic ataxia (predominant pyramidal signs) | Mild hyperintense pyramidal tracts; spinal cord atrophy | GJC2/SPG44 (NM_020435) | c.219_220delCC (p.L74fs*33) + c.254T > C/(p.V85A) |
| #8 | 20 | 32 | AR | Complex HSP + glaucoma | OPCA + medulla oblongata atrophy | SYNE1/SCAR8 NM_182961 | c.15049C > T/p.Q5017* (Homozygous) |
| #9 | 13 | 56 | AR | Severe spastic ataxia | normal | TTPA/AVED NM_000370D | c.553-1G > T (Homozygous) |
| #10 [31,37] | 41 | 54 | SP | Progressive spastic ataxia + psychosis; cognitive decline | Cortical atrophy; CA. Posterior leukoencephal | TUBB4A NM_006087 | c.545C > G/p.P182R (de novo) |
| #11 [32] | 30 | 42 | SP | Gait disorder, axial and limbs dystonic tremor | CA | PRKCG/SCA14 NM_002739.4; NM_001316329.1 | c.380A > C/p.Q127P (de novo) |
| #12 [36] | 44 | 48 | AD | Mild cerebellar ataxia and dysarthria | Severe CA | STUB1/SCA48 NM_001005920.2 | c.673C > T/p.Arg225* |
| #13 [36] | 51 | 54 | SP | Cerebellar ataxia and cognitive decline | Severe CA | STUB1/SCA48 NM_001005920.2 | c.721C > T/p.Arg241Trp |
| #14 [38] | 38 | 46 | AD | Gait ataxia + cognitive impairment | CA | TMEM240/SCA21 NM_001114748.2 | c.509C > T/p.P170L [36] |
| #15 [39] | 60 | 70 | SP | Pure AC | CA | KIF1B/CMT2A1 NM_015074.3 | c.3845C > G/pA1282G |
| #16 | 16 | 62 | SP | Ataxic gait and dysarthria | CA | PRKCG/SCA14 NM_002739.4; NM_001316329.1 | c.1928T > G/p.F643C |
| References | Subjects | Diagnostic Rate HSP | Diagnostic Rate CA |
|---|---|---|---|
| Our data TGP | 32 | 50% | 29.2% |
| D’Amore et al. 2018 [30] TGP | 239 | 29% | |
| Nemeth et al. 2013 [41] TGP | 50 | 18% | |
| Coutelier et al. 2018 [43] TES | 319 | 28.5% | |
| Lu et al. 2018 [44] TGP | 55 | 61.8% | |
| Burguez et al. 2017 [8] TGP | 29 | 48.3% | |
| Lynch et al. 2016 [45] TES + TGP | 40 | 52.5% | |
| Kara et al. 2016 [46] TES + WES | 97 | 49% | |
| Schule et al. 2016 [11] TGP + WES | 608 | 46% | |
| Fogel et al. 2014 [28] TES | 76 | 21% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riso, V.; Rossi, S.; Nicoletti, T.F.; Tessa, A.; Travaglini, L.; Zanni, G.; Aiello, C.; Perna, A.; Barghigiani, M.; Pomponi, M.G.; et al. Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience. Brain Sci. 2021, 11, 246. https://doi.org/10.3390/brainsci11020246
Riso V, Rossi S, Nicoletti TF, Tessa A, Travaglini L, Zanni G, Aiello C, Perna A, Barghigiani M, Pomponi MG, et al. Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience. Brain Sciences. 2021; 11(2):246. https://doi.org/10.3390/brainsci11020246
Chicago/Turabian StyleRiso, Vittorio, Salvatore Rossi, Tommaso F. Nicoletti, Alessandra Tessa, Lorena Travaglini, Ginevra Zanni, Chiara Aiello, Alessia Perna, Melissa Barghigiani, Maria Grazia Pomponi, and et al. 2021. "Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience" Brain Sciences 11, no. 2: 246. https://doi.org/10.3390/brainsci11020246
APA StyleRiso, V., Rossi, S., Nicoletti, T. F., Tessa, A., Travaglini, L., Zanni, G., Aiello, C., Perna, A., Barghigiani, M., Pomponi, M. G., Santorelli, F. M., & Silvestri, G. (2021). Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience. Brain Sciences, 11(2), 246. https://doi.org/10.3390/brainsci11020246