
A Core Transcription Regulatory Circuitry Defining Microglia Cell Identity Inferred from the Reanalysis of Multiple Human Microglia Differentiation Protocols


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials & Methods
2.1. Public Transcriptomes
2.2. Data Pre-Processing
2.3. Principal Component Analysis
2.4. Differential Gene Expression Analysis
2.5. Reconstruction of Gene Co-Regulatory Networks
3. Results
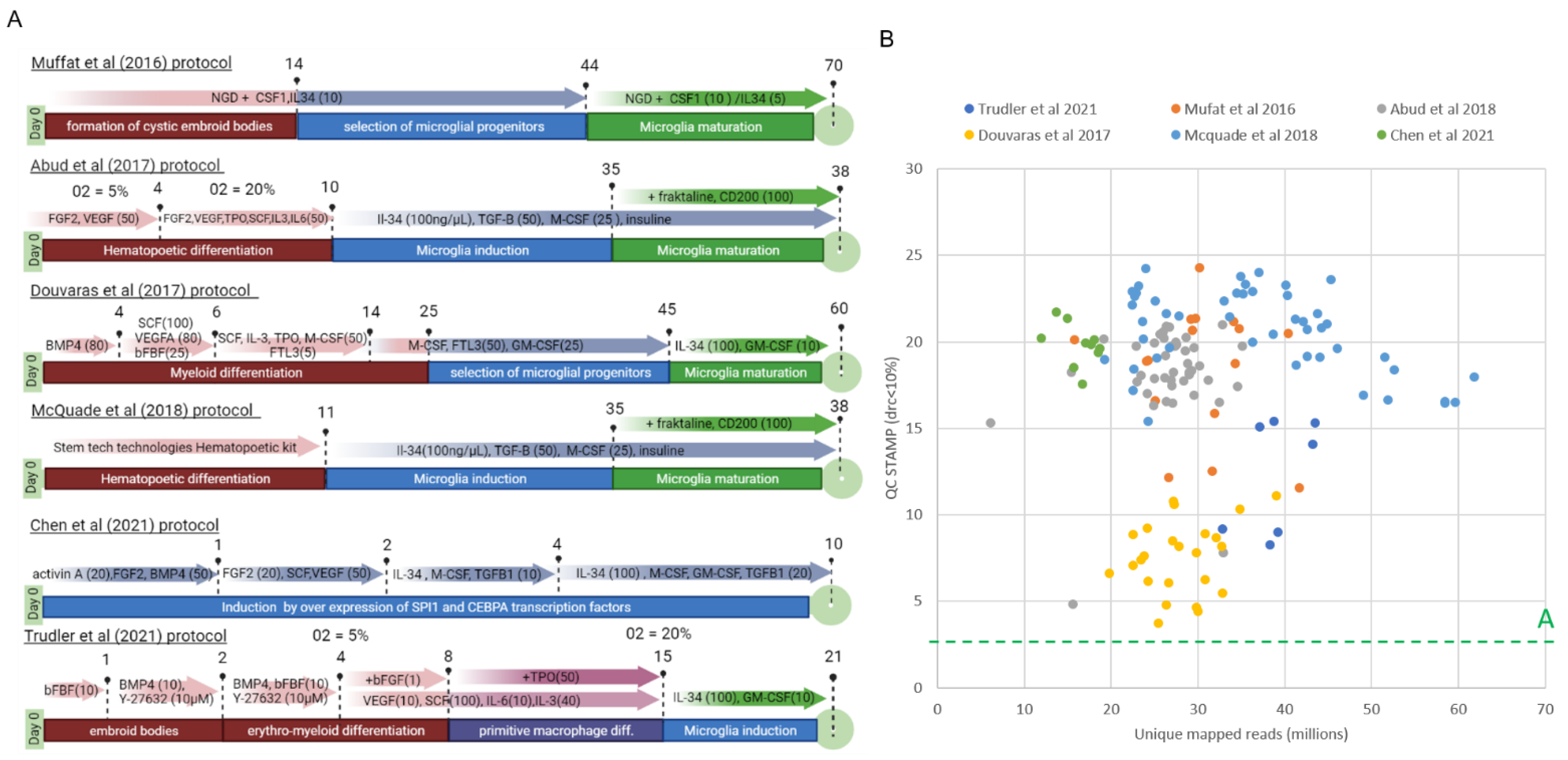
3.1. Comparative Reanalysis of Whole Transcriptome Readouts Generated within Six iPSC–Derived Microglia (iMGL) Differentiation Studies
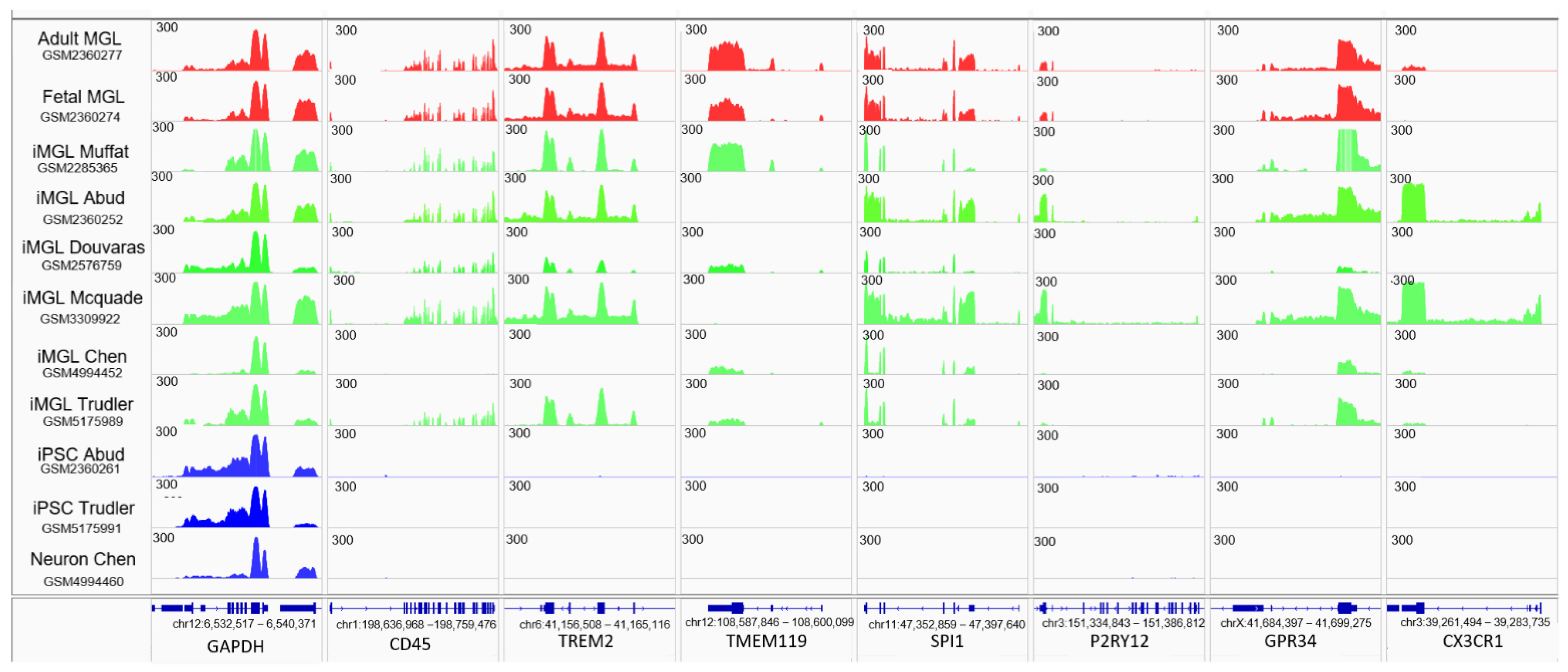
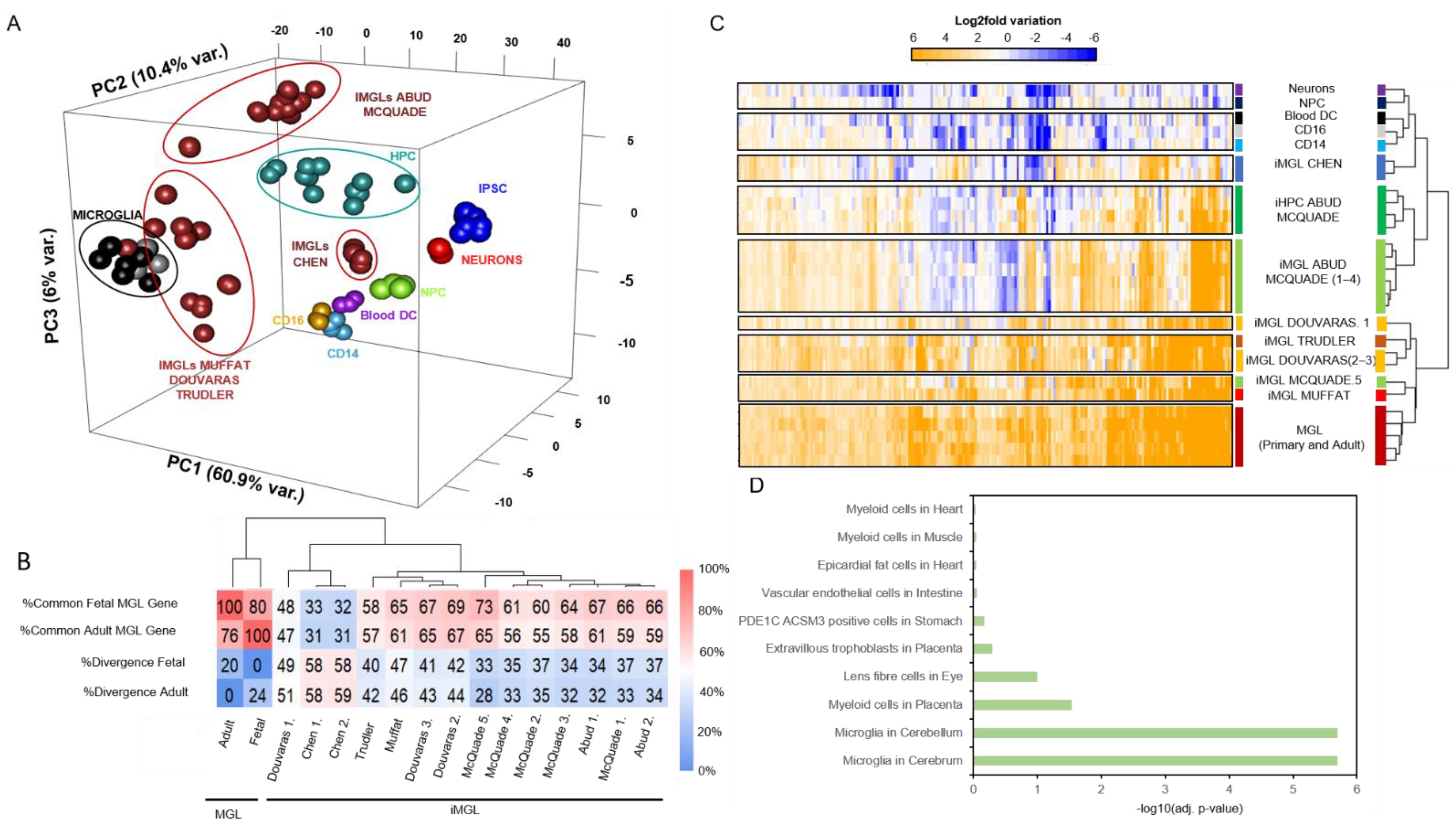
3.2. iMGL Transcriptome Datasets Issued from Different Protocols Present Variable Degree of Similarity with Control Microglia Samples
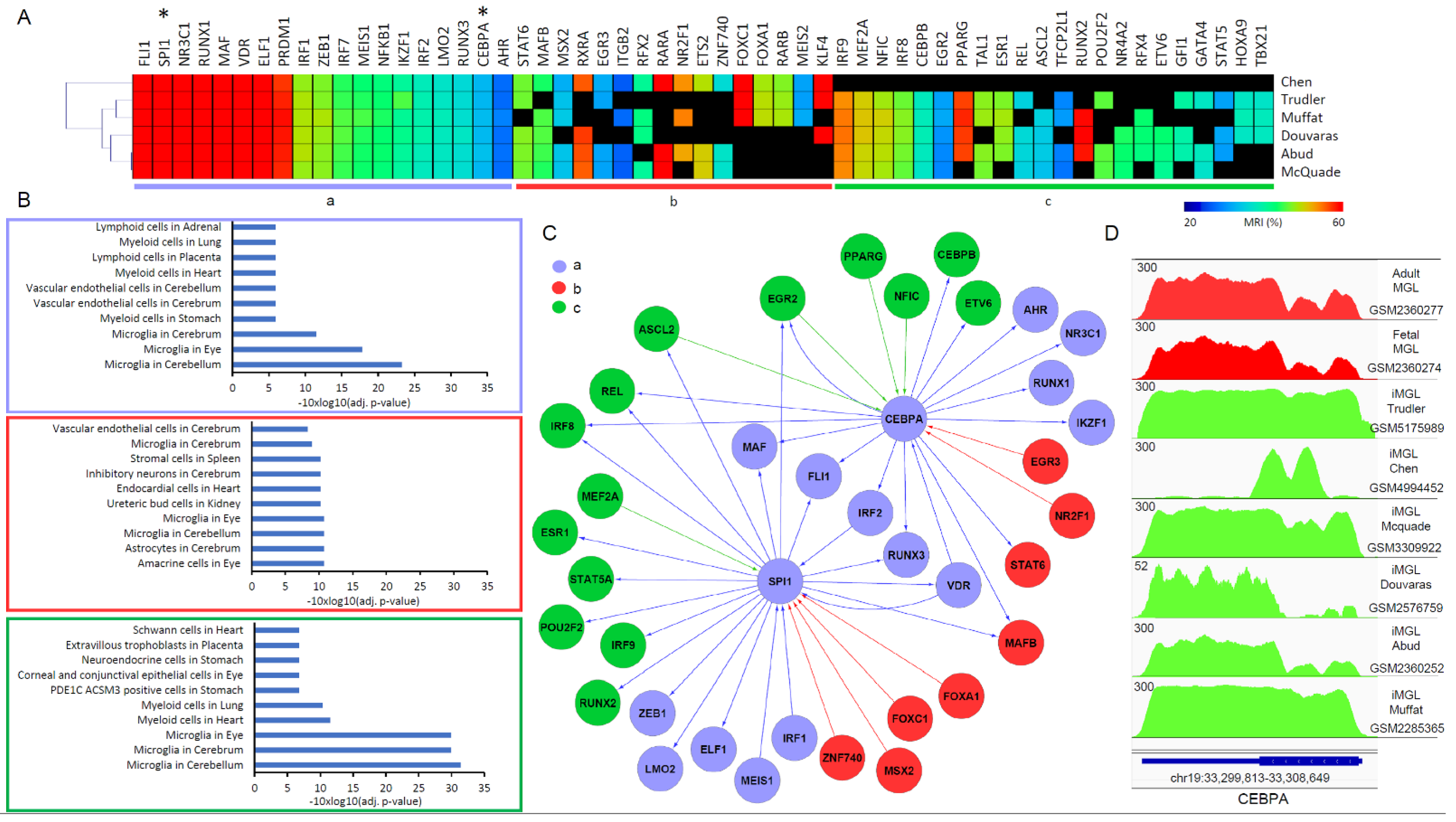
3.3. A Master TF Co-Regulatory Network Driving Microglia Differentiation Reveal Strengths and Weaknesses of the Compared Protocols
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sousa, C.; Biber, K.; Michelucci, A. Cellular and molecular characterization of microglia: A unique immune cell population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Tong, C.K.; Vidyadaran, S. Role of microglia in embryonic neurogenesis. Exp. Biol. Med. 2016, 241, 1669–1675. [Google Scholar] [CrossRef]
- Luo, X.; Chen, S.-D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Ferrer, I.J.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T.; Mayes, P.; Kielian, M. Characterization of microglial responses to Staphylococcus aureus: Effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J. Neuroimmunol. 2002, 130, 86–99. [Google Scholar] [CrossRef]
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflammation 2005, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Michell-Robinson, M.; Touil, H.; Healy, L.; Owen, D.; Durafourt, B.; Bar-Or, A.; Antel, J.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef]
- Wojtera, M.; Sikorska, B.; Sobow, T.; Liberski, P.P. Microglial cells in neurodegenerative disorders. Folia Neuropathol. 2005, 43, 311–321. [Google Scholar]
- Cho, S.-H.; Sun, B.; Zhou, Y.; Kauppinen, T.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 Protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef]
- De Pablos, R.M.; Herrera, A.J.; Espinosa-Oliva, A.M.; Sarmiento, M.; Muñoz, M.F.; Machado, A.; Venero, J.L. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J. Neuroinflamm. 2014, 11, 34. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Chagas, L.; Sandre, P.C.; Ribeiro, N.C.A.R.; Marcondes, H.; Silva, P.O.; Savino, W.; Serfaty, C.A. Environmental signals on microglial function during brain development, neuroplasticity, and disease. Int. J. Mol. Sci. 2020, 21, 2111. [Google Scholar] [CrossRef] [PubMed]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Di Paolo, T.; Tremblay, M. Microglial implication in Parkinson’s disease: Loss of Beneficial physiological roles or gain of inflammatory functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Microglial Activation and Inflammation as a Factor in the Pathogenesis of Progressive Supranuclear Palsy (PSP). Front. Neurosci. 2020, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- Koziorowski, D.; Figura, M.; Milanowski, Ł.M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of neurodegeneration in various forms of parkinsonism—Similarities and differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Li, S.; Cao, Q.; Wang, X.; Yan, Q.; Tu, X.; Zhu, Y.; Zhu, F. Human endogenous retrovirus W env increases nitric oxide production and enhances the migration ability of microglia by regulating the expression of inducible nitric oxide synthase. Virol. Sin. 2017, 32, 216–225. [Google Scholar] [CrossRef]
- Gruchot, J.; Kremer, D.; Küry, P. Neural cell responses upon exposure to human endogenous retroviruses. Front. Genet. 2019, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.; Elkhaloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Hänseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.; Chintawar, S.; Schnell, C.; Antel, J.; Allen, N.; et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.; Nguyen, C.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.; Marsh, S.; Fimbres, C.; et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Coburn, M.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein–antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef]
- Chen, S.-W.; Hung, Y.-S.; Fuh, J.-L.; Chen, N.-J.; Chu, Y.-S.; Chen, S.-C.; Fann, M.-J.; Wong, Y.-H. Efficient conversion of human induced pluripotent stem cells into microglia by defined transcription factors. Stem Cell Rep. 2021, 16, 1363–1380. [Google Scholar] [CrossRef]
- Davis, T.L.; Rebay, I. Master regulators in development: Views from the Drosophila retinal determination and mammalian pluripotency gene networks. Dev. Biol. 2017, 421, 93–107. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Cholley, P.-E.; Malysheva, V.; Nicaise, S.; Moehlin, J.; Gronemeyer, H.; Mendoza-Parra, M.A. A comprehensive resource for retrieving, visualizing, and integrating functional genomics data. Life Sci. Alliance 2020, 3, e201900546. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Parra, M.A.; Saleem, M.-A.M.; Blum, M.; Cholley, P.-E.; Gronemeyer, H. NGS-QC generator: A quality control system for ChIP-seq and related deep sequencing-generated datasets. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 243–265. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Cholley, P.-E.; Moehlin, J.; Rohmer, A.; Zilliox, V.; Nicaise, S.; Gronemeyer, H.; Mendoza-Parra, M.A. Modeling gene-regulatory networks to describe cell fate transitions and predict master regulators. NPJ Syst. Biol. Appl. 2018, 4, 29. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef]
- Banerjee, A.; Lu, Y.; Do, K.; Mize, T.; Wu, X.; Chen, X.; Chen, J. Validation of induced microglia-like cells (iMG Cells) for future studies of brain diseases. Front. Cell. Neurosci. 2021, 15, 85. [Google Scholar] [CrossRef]
- Grubman, A.; Vandekolk, T.H.; Schroeder, J.; Sun, G.; Hatwell-Humble, J.; Chan, J.; Oksanen, M.; Lehtonen, S.; Hunt, C.; Koistinaho, J.E.; et al. A CX3CR1 reporter hESC line facilitates integrative analysis of in-vitro-derived microglia and improved microglia identity upon neuron-glia co-culture. Stem Cell Rep. 2020, 14, 1018–1032. [Google Scholar] [CrossRef]
- Mendoza-Parra, M.-A.; Van Gool, W.; Saleem, M.A.M.; Ceschin, D.G.; Gronemeyer, H. A quality control system for profiles obtained by ChIP sequencing. Nucleic Acids Res. 2013, 41, e196. [Google Scholar] [CrossRef][Green Version]
- Masuda, T.; Tsuda, M.; Yoshinaga, R.; Tozaki-Saitoh, H.; Ozato, K.; Tamura, T.; Inoue, K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012, 1, 334–340. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial physiology and pathophysiology: Insights from genome-wide transcriptional profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular specificity of NF-κB Function in the nervous system. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Luna-Medina, R.; Sanz-SanCristobal, M.; Alvarez-Barrientos, A.; Santos, A.; Perez-Castillo, A. CCAAT/enhancer binding protein β deficiency provides cerebral protection following excitotoxic injury. J. Cell Sci. 2008, 121, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Hock, H.; Shimamura, A. ETV6 in hematopoiesis and leukemia predisposition. Semin. Hematol. 2017, 54, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Stence, N.; Waite, M.; Dailey, M. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef]
- Sievers, J.; Parwaresch, R.; Wottge, H.-U. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: Morphology. Glia 1994, 12, 245–258. [Google Scholar] [CrossRef]
- Hinze, A.; Stolzing, A. Microglia differentiation using a culture system for the expansion of mice non-adherent bone marrow stem cells. J. Inflamm. 2012, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.; Ousterout, D.G.; Leong, K.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, A.; Stüder, F.; Colombo, B.M.; Mendoza-Parra, M.A. A Core Transcription Regulatory Circuitry Defining Microglia Cell Identity Inferred from the Reanalysis of Multiple Human Microglia Differentiation Protocols. Brain Sci. 2021, 11, 1338. https://doi.org/10.3390/brainsci11101338
Aubert A, Stüder F, Colombo BM, Mendoza-Parra MA. A Core Transcription Regulatory Circuitry Defining Microglia Cell Identity Inferred from the Reanalysis of Multiple Human Microglia Differentiation Protocols. Brain Sciences. 2021; 11(10):1338. https://doi.org/10.3390/brainsci11101338
Chicago/Turabian StyleAubert, Antoine, François Stüder, Bruno Maria Colombo, and Marco Antonio Mendoza-Parra. 2021. "A Core Transcription Regulatory Circuitry Defining Microglia Cell Identity Inferred from the Reanalysis of Multiple Human Microglia Differentiation Protocols" Brain Sciences 11, no. 10: 1338. https://doi.org/10.3390/brainsci11101338
APA StyleAubert, A., Stüder, F., Colombo, B. M., & Mendoza-Parra, M. A. (2021). A Core Transcription Regulatory Circuitry Defining Microglia Cell Identity Inferred from the Reanalysis of Multiple Human Microglia Differentiation Protocols. Brain Sciences, 11(10), 1338. https://doi.org/10.3390/brainsci11101338