Distant Organ Damage in Acute Brain Injury



Abstract
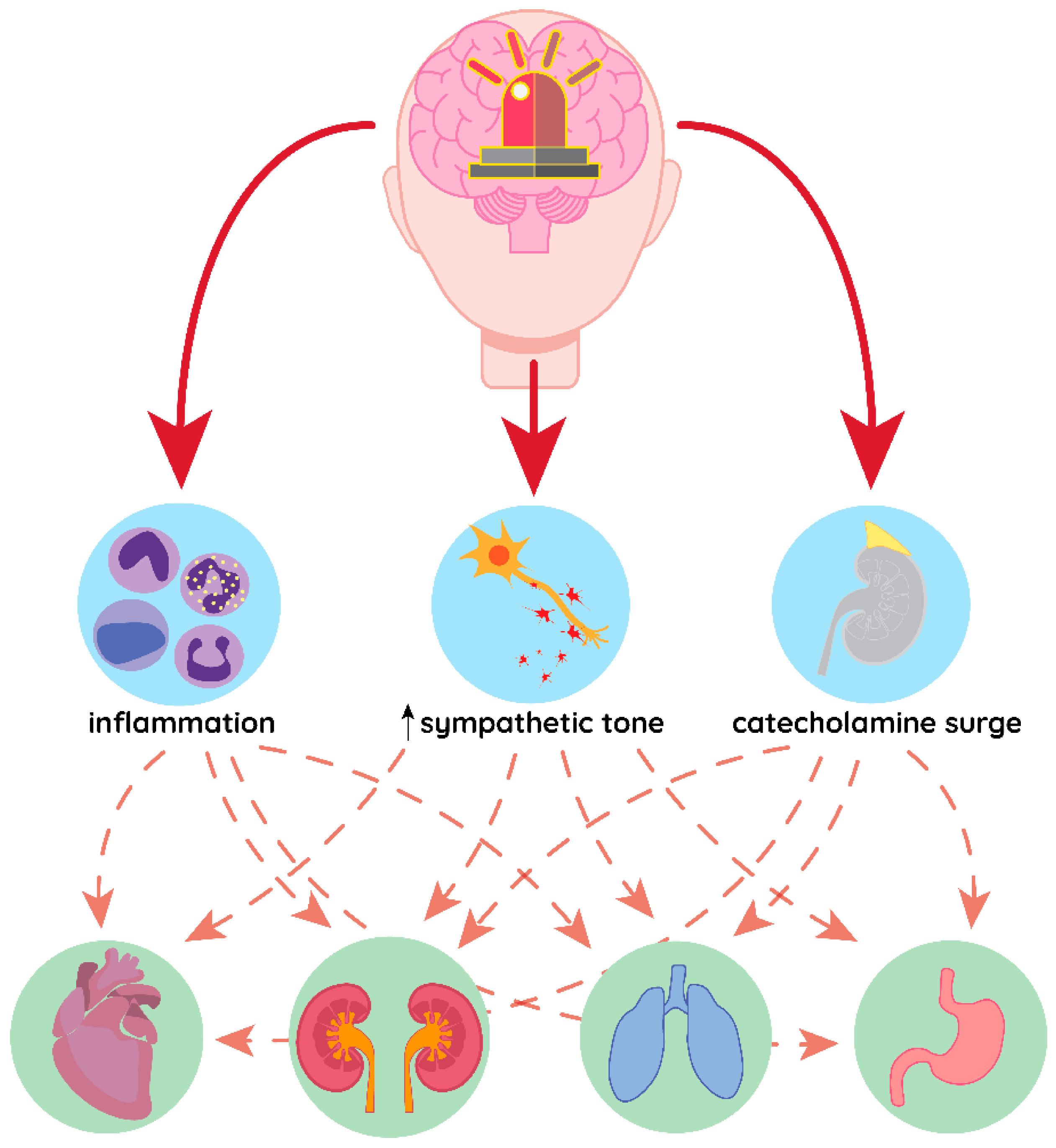
1. Introduction
2. Endocrine Manifestations of Brain Injury
2.1. Pituitary Dysfunction
2.2. Antidiuretic Hormone Secretion Abnormalities
2.3. Thyroid Hormone
2.4. Adrenal Cortex Hormones
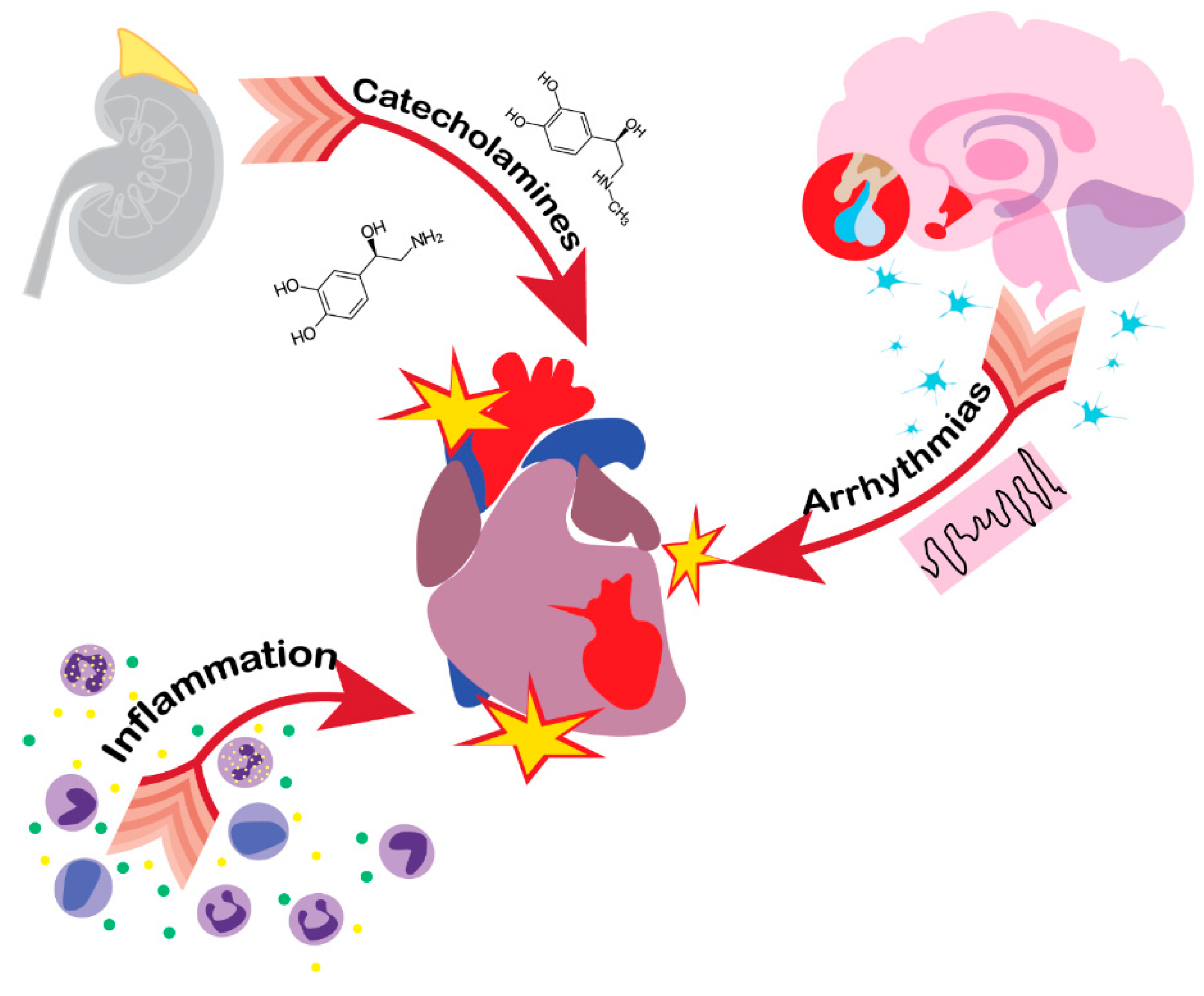
3. Heart
4. Lungs
5. Kidneys
6. Immune System
7. Digestive Tract
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Causes of Death. Available online: https://www.who.int/data/gho/data/themes/topics/causes-of-death (accessed on 13 November 2020).
- Sharma, R.; Shultz, S.R.; Robinson, M.J.; Belli, A.; Hibbs, M.L.; O’Brien, T.J.; Semple, B.D. Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav. Immun. 2019, 79, 63–74. [Google Scholar] [PubMed]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [PubMed]
- Yang, W.H.; Chen, P.C.; Wang, T.C.; Kuo, T.Y.; Cheng, C.Y.; Yang, Y.H. Endocrine dysfunction following traumatic brain injury: A 5-year follow-up nationwide-based study. Sci. Rep. 2016, 6, 32987. [Google Scholar] [PubMed]
- Agrawal, A.; Reddy, P.A.; Prasad, N.R. Endocrine manifestations of traumatic brain injury. Indian J. Neurotrauma 2012, 9, 123–128. [Google Scholar]
- Kelly, D.F.; Gonzalo, I.T.; Cohan, P.; Berman, N.; Swerdloff, R.; Wang, C. Hypopituitarism following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A preliminary report. J. Neurosurg. 2000, 93, 743–752. [Google Scholar]
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar]
- Glynn, N.; Agha, A. The frequency and the diagnosis of pituitary dysfunction after traumatic brain injury. Pituitary 2019, 22, 249–260. [Google Scholar]
- Tanriverdi, F.; Unluhizarci, K.; Kelestimur, F. Pituitary function in subjects with mild traumatic brain injury: A review of literature and proposal of a screening strategy. Pituitary 2010, 13, 146–153. [Google Scholar]
- Gray, S.; Bilski, T.; Dieudonne, B.; Saeed, S. Hypopituitarism after Traumatic Brain Injury. Cureus 2019, 11, 4163. [Google Scholar]
- Booij, H.A.; Gaykema, W.D.C.; Kuijpers, K.A.J.; Pouwels, M.J.M.; den Hertog, H.M. Pituitary dysfunction and association with fatigue in stroke and other acute brain injury. Endocr. Connect. 2018, 7, 223–237. [Google Scholar]
- Bondanelli, M.; Ambrosio, M.R.; Zatelli, M.C.; Basaglia, N.; Degli Uberti, E.C. Prevalence of hypopituitarism in patients with cerebrovascular diseases. J. Endocrinol. Investig. 2008, 31, 16–20. [Google Scholar]
- Aimaretti, G.; Ambrosio, M.R.; di Somma, C.; Fusco, A.; Cannavò, S.; Gasperi, M.; Scaroni, C.; de Marinis, L.; Benvenga, S.; Uberti, E.C.; et al. Traumatic brain injury and subarachnoid haemorrhage are conditions at high risk for hypopituitarism: Screening study at 3 months after the brain injury. Clin. Endocrinol. 2004, 61, 320–326. [Google Scholar]
- Bondanelli, M.; de Marinis, L.; Ambrosio, M.R.; Monesi, M.; Valle, D.; Zatelli, M.C.; Fusco, A.; Bianchi, A.; Farneti, M.; Uberti, E.C.I.D. Occurrence of pituitary dysfunction following traumatic brain injury. J. Neurotrauma 2004, 21, 685–696. [Google Scholar] [PubMed]
- Powner, D.J.; Boccalandro, C.; Alp, M.S.; Vollmer, D.G. Endocrine failure after traumatic brain injury in adults. Neurocrit. Care 2006, 5, 61–70. [Google Scholar] [PubMed]
- Agha, A.; Phillips, J.; O’Kelly, P.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic hypopituitarism: Implications for assessment and treatment. Am. J. Med. 2005, 118, 1416. [Google Scholar]
- Klose, M.; Feldt-Rasmussen, U. Does the type and severity of brain injury predict hypothalamo-pituitary dysfunction? Does post-traumatic hypopituitarism predict worse outcome? Pituitary 2008, 11, 255–261. [Google Scholar]
- Khajeh, L.; Blijdorp, K.; Neggers, S.J.; Ribbers, G.M.; Dippel, D.W.; van Kooten, F. Hypopituitarism after subarachnoid haemorrhage, do we know enough? BMC Neurol. 2014, 14, 205. [Google Scholar]
- Masel, B.E.; Urban, R. Chronic Endocrinopathies in Traumatic Brain Injury Disease. J. Neurotrauma 2015, 32, 1902–1910. [Google Scholar]
- Kgosidialwa, O.; Agha, A. Hypopituitarism post traumatic brain injury (TBI): Review. Ir. J. Med. Sci. 2019, 188, 1201–1206. [Google Scholar]
- Dusick, J.R.; Wang, C.; Cohan, P.; Swerdloff, R.; Kelly, D.F. Pathophysiology of hypopituitarism in the setting of brain injury. Pituitary 2012, 15, 2–9. [Google Scholar]
- Maiya, B.; Newcombe, V.; Nortje, J.; Bradley, P.; Bernard, F.; Chatfield, D.; Outtrim, J.; Hutchinson, P.; Matta, B.; Antoun, N.; et al. Magnetic resonance imaging changes in the pituitary gland following acute traumatic brain injury. Intensive Care Med. 2008, 34, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, R.N.; Fisher, R.S. Pituitary lesions in craniocerebral injuries. Arch. Pathol. 1969, 88, 242–248. [Google Scholar] [PubMed]
- Barugh, A.J.; Gray, P.; Shenkin, S.D.; MacLullich, A.M.; Mead, G.E. Cortisol levels and the severity and outcomes of acute stroke: A systematic review. J. Neurol. 2014, 261, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Ntali, G.; Tsagarakis, S. Pituitary dysfunction after traumatic brain injury: Prevalence and screening strategies. Expert Rev. Endocrinol. Metab. 2020, 15, 341–354. [Google Scholar] [CrossRef]
- Capatina, C.; Paluzzi, A.; Mitchell, R.; Karavitaki, N. Diabetes Insipidus after Traumatic Brain Injury. J. Clin. Med. 2015, 4, 1448–1462. [Google Scholar] [CrossRef]
- Cui, H.; He, G.; Yang, S.; Lv, Y.; Jiang, Z.; Gang, X.; Wang, G. Inappropriate Antidiuretic Hormone Secretion and Cerebral Salt-Wasting Syndromes in Neurological Patients. Front. Neurosci. 2019, 13, 1170. [Google Scholar] [CrossRef]
- Jones, D.P. Syndrome of Inappropriate Secretion of Antidiuretic Hormone and Hyponatremia. Pediatr. Rev. 2018, 39, 27–35. [Google Scholar] [CrossRef]
- Mangieri, P.; Suzuki, K.; Ferreira, M.; Domingues, L.; Casulari, L.A. Evaluation of pituitary and thyroid hormones in patients with subarachnoid hemorrhage due to ruptured intracranial aneurysm. Arq. Neuro-Psiquiatr. 2003, 61, 14–19. [Google Scholar] [CrossRef]
- Gaddam, S.S.; Buell, T.; Robertson, C.S. Systemic manifestations of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 205–218. [Google Scholar]
- Euthyroid Sick Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482219/ (accessed on 13 November 2020).
- Slag, M.F.; Morley, J.E.; Elson, M.K.; Crowson, T.W.; Nuttall, F.Q.; Shafer, R.B. Hypothyroxinemia in critically ill patients as a predictor of high mortality. JAMA 1981, 245, 43–45. [Google Scholar] [CrossRef]
- Alevizaki, M.; Synetou, M.; Xynos, K.; Pappa, T.; Vemmos, K.N. Low triiodothyronine: A strong predictor of outcome in acute stroke patients. Eur. J. Clin. Investig. 2007, 37, 651–657. [Google Scholar] [CrossRef]
- O’Keefe, L.M.; Conway, S.E.; Czap, A.; Malchoff, C.D.; Benashski, S.; Fortunato, G.; Staff, I.; McCullough, L.D. Thyroid hormones and functional outcomes after ischemic stroke. Thyroid Res. 2015, 4, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Klose, M.; Vita, R.; Feldt-Rasmussen, U. Less known aspects of central hypothyroidism: Part 1—Acquired etiologies. J. Clin. Transl. Endocrinol. 2018, 14, 25–33. [Google Scholar] [CrossRef]
- Woolf, P.D.; Lee, L.A.; Hamill, R.W.; McDonald, J.V. Thyroid test abnormalities in traumatic brain injury: Correlation with neurologic impairment and sympathetic nervous system activation. Am. J. Med. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Hwang, J.J.; Hwang, D.Y. Treatment of endocrine disorders in the neuroscience intensive care unit. Curr. Treat Options Neurol. 2014, 16, 271. [Google Scholar] [CrossRef]
- Fliers, E.; Bianco, A.C.; Langouche, L.; Boelen, A. Thyroid function in critically ill patients. Lancet Diabetes Endocrinol. 2015, 3, 816–825. [Google Scholar] [CrossRef]
- Powner, D.J.; Boccalandro, C. Adrenal insufficiency following traumatic brain injury in adults. Curr. Opin. Crit. Care 2008, 14, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Swaminathan, G.; Nair, S.; Joseph, M. Hyponatremia in Traumatic Brain Injury: A Practical Management Protocol. World Neurosurg. 2017, 108, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Sébille, V.; Charpentier, C.; Bollaert, P.E.; François, B.; Korach, J.M.; Capellier, G.; Cohen, Y.; Azoulay, E.; Troché, G.; et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002, 288, 862–871. [Google Scholar] [CrossRef]
- Cohan, P.; Wang, C.; McArthur, D.L.; Cook, S.W.; Dusick, J.R.; Armin, B.B.; Swerdloff, R.; Vespa, P.; Muizelaar, J.; Cryer, H.G.; et al. Acute secondary adrenal insufficiency after traumatic brain injury: A prospective study. Crit. Care Med. 2005, 33, 2358–2366. [Google Scholar] [CrossRef]
- Kleindienst, A.; Brabant, G.; Bock, C.; Maser-Gluth, C.; Buchfelder, M. Neuroendocrine function following traumatic brain injury and subsequent intensive care treatment: A prospective longitudinal evaluation. J. Neurotrauma 2009, 26, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Yeliosof, O.; Gangat, M. Diagnosis and management of hypopituitarism. Curr. Opin. Pediatr. 2019, 31, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Stewart, P.M. Diagnosis and Treatment of ACTH Deficiency. Rev. Endocr. Metab. Disord. 2005, 6, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kgosidialwa, O.; Hakami, O.; Muhammad Zia-Ul-Hussnain, H.; Agha, A. Growth Hormone Deficiency Following Traumatic Brain Injury. Int. J. Mol. Sci. 2019, 20, 3323. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, R.; Zylberstejn, D.S.; Esteves, S.C. Hypogonadotropic hypogonadism revisited. Clinics 2013, 68, 81–88. [Google Scholar] [CrossRef]
- High, W.M., Jr.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Chinga-Alayo, E.; Villena, J.; Evans, A.T.; Zimic, M. Thyroid hormone levels improve the prediction of mortality among patients admitted to the intensive care unit. Intensive Care Med. 2005, 31, 1356–1361. [Google Scholar] [CrossRef]
- Cushing, H. Concerning a definite regulatory mechanism of the vasomotor centre which controls blood pressure during cerebral compression. Bull. Johns Hopkins Hosp. 1901, 12, 290–292. [Google Scholar]
- Ay, H.; Koroshetz, W.J.; Benner, T.; Vangel, M.G.; Melinosky, C.; Arsava, E.M.; Ayata, C.; Zhu, M.; Schwamm, L.H.; Sorensen, A.G. Neuroanatomic correlates of stroke-related myocardial injury. Neurology 2006, 66, 1325–1329. [Google Scholar] [CrossRef]
- Jachuck, S.J.; Ramani, P.S.; Clark, F.; Kalbag, R.M. Electrocardiographic abnormalities associated with raised intracranial pressure. Br. Med. J. 1975, 1, 242–244. [Google Scholar] [CrossRef]
- Rem, J.A.; Hachinski, V.C.; Boughner, D.R.; Barnett, H.J. Value of cardiac monitoring and echocardiography in TIA and stroke patients. Stroke 1985, 16, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Venkata, C.; Kasal, J. Cardiac Dysfunction in Adult Patients with Traumatic Brain Injury: A Prospective Cohort Study. Clin. Med. Res. 2018, 16, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sijercic, S.; Krdzalic, A.; Avdagic, H.; Krdzalic, G. Incidence of Cardiac Dysfunction after Brain Injury. Med. Arch. 2018, 72, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Naidech, A.M.; Kreiter, K.T.; Janjua, N.; Ostapkovich, N.D.; Parra, A.; Commichau, C.; Fitzsimmons, B.M.; Connolly, E.S.; Mayer, S.A. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005, 112, 2851–2856. [Google Scholar] [CrossRef] [PubMed]
- Van der Bilt, I.A.; Hasan, D.; Vandertop, W.P.; Wilde, A.A.M.; Algra, A.; Visser, F.; Rinkel, G.J.E. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: A meta-analysis. Neurology 2009, 72, 635–642. [Google Scholar] [CrossRef]
- Zygun, D.A.; Kortbeek, J.B.; Fick, G.H.; Laupland, K.B.; Doig, C.J. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit. Care Med. 2005, 33, 654–660. [Google Scholar] [CrossRef]
- Touzé, E.; Varenne, O.; Chatellier, G.; Peyrard, S.; Rothwell, P.M.; Mas, J.L. Risk of myocardial infarction and vascular death after transient ischemic attack and ischemic stroke: A systematic review and meta-analysis. Stroke 2005, 36, 2748–2755. [Google Scholar] [CrossRef]
- Sato, K.; Masuda, T.; Izumi, T. Subarachnoid hemorrhage and myocardial damage clinical and experimental studies. Jpn. Heart J. 1999, 40, 683–701. [Google Scholar] [CrossRef]
- Junttila, E.; Vaara, M.; Koskenkari, J.; Ohtonen, P.; Karttunen, A.; Raatikainen, P.; Ala-Kokko, T. Repolarization abnormalities in patients with subarachnoid and intracerebral hemorrhage: Predisposing factors and association with outcome. Anesth. Analg. 2013, 116, 190–197. [Google Scholar] [CrossRef]
- Li, W.; Li, L.; Chopp, M.; Venkat, P.; Zacharek, A.; Chen, Z.; Landschoot-Ward, J.; Yan, T.; Chen, J. Intracerebral Hemorrhage Induces Cardiac Dysfunction in Mice Without Primary Cardiac Disease. Front. Neurol. 2018, 9, 965. [Google Scholar] [CrossRef]
- Dias, V.; Cabral, S.; Meireles, A.; Gomes, C.; Antunes, N.; Vieira, M.; Caiado, L.; Torres, S. Stunned myocardium following ischemic stroke. Case report. Cardiology 2009, 113, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Wybraniec, M.T.; Mizia-Stec, K.; Krzych, Ł. Neurocardiogenic injury in subarachnoid hemorrhage: A wide spectrum of catecholamin-mediated brain-heart interactions. Cardiol. J. 2014, 21, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tung, P.; Kopelnik, A.; Banki, N.; Ong, K.; Ko, N.; Lawton, M.T.; Gress, D.; Drew, B.; Foster, E.; Parmley, W.; et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004, 35, 548–551. [Google Scholar] [CrossRef]
- Salem, R.; Vallée, F.; Dépret, F.; Callebert, J.; Maurice, J.P.S.; Marty, P.; Matéo, J.; Madadaki, C.; Houdart, E.; Bresson, D.; et al. Subarachnoid hemorrhage induces an early and reversible cardiac injury associated with catecholamine release: One-week follow-up study. Crit. Care 2014, 18, 558. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Norris, J.W.; Hachniski, V.C.; Sole, M. Plasma norepinephrine in stroke. Stroke 1981, 12, 200–204. [Google Scholar] [CrossRef]
- Naredi, S.; Lambert, G.; Edén, E.; Zäll, S.; Runnerstam, M.; Rydenhag, B.; Friberg, P. Increased sympathetic nervous activity in patients with nontraumatic subarachnoid hemorrhage. Stroke 2000, 31, 901–906. [Google Scholar] [CrossRef]
- Sander, D.; Winbeck, K.; Klingelhöfer, J.; Etgen, T.; Conrad, B. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology 2001, 57, 833–838. [Google Scholar] [CrossRef]
- Hawkins, W.E.; Clower, B.R. Myocardial damage after head trauma and simulated intracranial haemorrhage in mice: The role of the autonomic nervous system. Cardiovasc. Res. 1971, 5, 524–529. [Google Scholar] [CrossRef]
- Schömig, A. Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation 1990, 82, 13–22. [Google Scholar]
- Mertes, P.M.; Carteaux, J.P.; Jaboin, Y.; Pinelli, G.; El Abassi, K.; Dopff, C.; Atkinson, J.; Villemot, J.P.; Burlet, C.; Boulange, M. Estimation of myocardial interstitial norepinephrine release after brain death using cardiac microdialysis. Transplantation 1994, 57, 371–377. [Google Scholar] [CrossRef]
- Wallner, M.; Duran, J.M.; Mohsin, S.; Troupes, C.D.; Vanhoutte, D.; Borghetti, G.; Vagnozzi, R.J.; Gross, P.; Yu, D.; Trappanese, D.M.; et al. Acute Catecholamine Exposure Causes Reversible Myocyte Injury without Cardiac Regeneration. Circ. Res. 2016, 119, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Rona, G. Catecholamine cardiotoxicity. J. Mol. Cell. Cardiol. 1985, 17, 291–306. [Google Scholar] [CrossRef]
- Mann, D.L.; Kent, R.L.; Parsons, B.; Cooper, G., 4th. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 1992, 85, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, S.M. Neurogenic cardiac effects of cerebrovascular disease. Curr. Opin. Neurol. 2014, 7, 20–24. [Google Scholar] [CrossRef]
- Huang, C.C.; Huang, C.H.; Kuo, H.Y.; Chan, C.M.; Chen, J.H.; Chen, W.L. The 12-lead electrocardiogram in patients with subarachnoid hemorrhage: Early risk prognostication. Am. J. Emerg. Med. 2012, 30, 732–736. [Google Scholar] [CrossRef]
- Oppenheimer, S.M.; Gelb, A.; Girvin, J.P.; Hachinski, V.C. Cardiovascular effects of human insular cortex stimulation. Neurology 1992, 42, 1727–1732. [Google Scholar] [CrossRef]
- Colivicchi, F.; Bassi, A.; Santini, M.; Caltagirone, C. Prognostic implications of right-sided stroke with insular involvement. Stroke 2004, 35, 2094–2098. [Google Scholar] [CrossRef]
- Melville, K.; Blum, B.; Shister, H.; Silver, M. Cardiac Ischemic Changes and Arrhythmias Induced by Hypothalamic Stimulation. Am. J. Cardiol. 1963, 12, 781–791. [Google Scholar] [CrossRef]
- Chen, Z.; Venkat, P.; Seyfried, D.; Chopp, M.; Yan, T.; Chen, J. Brain-Heart Interaction: Cardiac Complications after Stroke. Circ. Res. 2017, 121, 451–468. [Google Scholar] [CrossRef]
- Eitel, I.; Lücke, C.; Grothoff, M.; Sareban, M.; Schuler, G.; Thiele, H.; Gutberlet, M. Inflammation in takotsubo cardiomyopathy: Insights from cardiovascular magnetic resonance imaging. Eur. Radiol. 2010, 20, 422–431. [Google Scholar] [CrossRef]
- Andò, G.; Trio, O.; de Gregorio, C. Catecholamine-induced stress cardiomyopathies: More similarities than differences. Int. J. Cardiol. 2013, 168, 4453–4454. [Google Scholar] [CrossRef] [PubMed]
- Guglin, M.; Novotorova, I. Neurogenic stunned myocardium and takotsubo cardiomyopathy are the same syndrome: A pooled analysis. Congest. Heart Fail. 2011, 17, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tateishi, H.; Uchida, T.; Dote, K.; Ishihara, M. Tako-Tsubo-like left ventricular dysfunction due to multivessel coronary spasm. In Clinical Aspect of Myocardial Injury: From Ischemia to Heart Failure; Kodama, K., Haze, K., Hori, M., Eds.; Kagakuhyoronsha Publishing Co.: Tokyo, Japan, 1990; pp. 56–64. (In Japanese) [Google Scholar]
- Scantlebury, D.C.; Prasad, A. Diagnosis of takotsubo cardiomyopathy—Mayo Clinic criteria. Circ. J. 2014, 78, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Ako, J.; Honda, Y.; Fitzgerald, P.J. Tako-tsubo-like left ventricular dysfunction. Circulation 2003, 108, 158. [Google Scholar] [CrossRef] [PubMed]
- Boland, T.A.; Lee, V.H.; Bleck, T.P. Stress-induced cardiomyopathy. Crit. Care Med. 2015, 43, 686–693. [Google Scholar] [CrossRef]
- Kato, K.; Lyon, A.R.; Ghadri, J.R.; Templin, C. Takotsubo syndrome: Aetiology, presentation and treatment. Heart 2017, 103, 1461–1469. [Google Scholar] [CrossRef]
- Bulsara, K.R.; McGirt, M.J.; Liao, L.; Villavicencio, A.T.; Borel, C.; Alexander, M.J.; Friedman, A.H. Use of the peak troponin value to differentiate myocardial infarction from reversible neurogenic left ventricular dysfunction associated with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2003, 98, 524–528. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Madhavan, M.; Prasad, A. Brain natriuretic peptide in apical ballooning syndrome (Takotsubo/stress cardiomyopathy): Comparison with acute myocardial infarction. Coron. Artery Dis. 2012, 23, 259–264. [Google Scholar] [CrossRef]
- Hurst, R.T.; Askew, J.W.; Reuss, C.S.; Lee, R.W.; Sweeney, J.P.; Fortuin, F.D.; Oh, J.K.; Tajik, A.J. Transient midventricular ballooning syndrome: A new variant. J. Am. Coll. Cardiol. 2006, 48, 579–583. [Google Scholar] [CrossRef]
- Cacciotti, L.; Camastra, G.S.; Musarò, S.; Proietti, I.; Semeraro, R.; Martina, C.; Lupparelli, F.; Ansalone, G. Stress cardiomyopathy: Transient basal ballooning. J. Cardiovasc. Med. 2010, 11, 764–767. [Google Scholar] [CrossRef]
- Mansencal, N.; Pellerin, D.; Lamar, A.; Beauchet, A.; El Mahmoud, R.; Pillière, R.; McKenna, W.J.; Dubourg, O. Diagnostic value of contrast echocardiography in Tako-Tsubo cardiomyopathy. Arch. Cardiovasc. Dis. 2010, 103, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Ghadri, J.; Wittstein, I.S.; Prasad, A.; Sharkey, S.; Dote, K.; Akashi, Y.J.; Cammann, V.L.; Crea, F.; Galiuto, L.; Desmet, W.; et al. International Expert Consensus Document on Takotsubo Syndrome (Part II): Diagnostic Workup, Outcome, and Management. Eur. Heart J. 2018, 39, 2047–2062. [Google Scholar] [CrossRef] [PubMed]
- Biso, S.; Wongrakpanich, S.; Agrawal, A.; Yadlapati, S.; Kishlyansky, M.; Figueredo, V. A Review of Neurogenic Stunned Myocardium. Cardiovasc. Psychiatry Neurol. 2017, 2017, 5842182. [Google Scholar] [CrossRef] [PubMed]
- Kerro, A.; Woods, T.; Chang, J.J. Neurogenic stunned myocardium in subarachnoid hemorrhage. J. Crit. Care 2017, 38, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Inamasu, J.; Watanabe, E.; Okuda, K.; Kumai, T.; Sugimoto, K.; Ozaki, Y.; Hirose, Y. Are there differences between Takotsubo cardiomyopathy and neurogenic stunned myocardium? A prospective observational study. Int. J. Cardiol. 2014, 177, 1108–1110. [Google Scholar] [CrossRef]
- Lyon, A.R.; Rees, P.S.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (Takotsubo) cardiomyopathy—A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 22–29. [Google Scholar] [CrossRef]
- Sugimoto, K.; Inamasu, J.; Hirose, Y.; Kato, Y.; Ito, K.; Iwase, M.; Sugimoto, K.; Watanabe, E.; Takahashi, A.; Ozaki, Y. The role of norepinephrine and estradiol in the pathogenesis of cardiac wall motion abnormality associated with subarachnoid hemorrhage. Stroke 2012, 43, 1897–1903. [Google Scholar] [CrossRef]
- Davison, D.L.; Terek, M.; Chawla, L.S. Neurogenic pulmonary edema. Crit. Care 2012, 16, 212. [Google Scholar] [CrossRef]
- Saracen, A.; Kotwica, Z.; Woźniak-Kosek, A.; Kasprzak, P. Neurogenic Pulmonary Edema in Aneurysmal Subarachnoid Hemorrhage. Adv. Exp. Med. Biol. 2016, 952, 35–39. [Google Scholar]
- Kimura, T.; Kamide, T.; Onodera, K.; Tabata, S.; Shibata, A.; Suzuki, K.; Takeda, R.; Ikeda, T.; Kikkawa, Y.; Iihoshi, S.; et al. Clinical Features of Neurogenic Pulmonary Edema in Patients with Subarachnoid Hemorrhage. World Neurosurg. 2020, 135, 505–509. [Google Scholar] [CrossRef]
- Bahloul, M.; Chaari, A.N.; Kallel, H.; Khabir, A.; Ayadi, A.; Charfeddine, H.; Hergafi, L.; Chaari, A.D.; Chelly, H.E.; Ben Hamida, C.; et al. Neurogenic pulmonary edema due to traumatic brain injury: Evidence of cardiac dysfunction. Am. J. Crit. Care 2006, 15, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.J.; Pittet, J.F.; Kerby, J.D.; Bosarge, P.L.; Wagener, B.M. Acute brain trauma, lung injury, and pneumonia: More than just altered mental status and decreased airway protection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; O’Kane, R. The Extracranial Consequences of Subarachnoid Hemorrhage. World Neurosurg. 2018, 109, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Naidech, A.M. Intracranial hemorrhage. Am. J. Respir. Crit. Care Med. 2011, 184, 998–1006. [Google Scholar] [CrossRef]
- Reuter-Rice, K.; Duthie, S.; Hamrick, J. Neurogenic pulmonary edema associated with pediatric status epilepticus. Pediatr. Emerg. Care 2011, 27, 957–958. [Google Scholar] [CrossRef]
- Sarı, M.Y.; Yıldızdaş, R.D.; Yükselmiş, U.; Horoz, Ö.Ö. Our patients followed up with a diagnosis of neurogenic pulmonary edema. Turk Pediatri Ars. 2015, 50, 241–244. [Google Scholar] [CrossRef]
- Manto, A.; De Gennaro, A.; Manzo, G.; Serino, A.; Quaranta, G.; Cancella, C. Early endovascular treatment of aneurysmal subarachnoid hemorrhage complicated by neurogenic pulmonary edema and Takotsubo-like cardiomyopathy. Neuroradiol. J. 2014, 27, 356–360. [Google Scholar] [CrossRef]
- Simon, R.P. Neurogenic pulmonary edema. Neurol. Clin. 1993, 11, 309–323. [Google Scholar] [CrossRef]
- Šedý, J.; Kuneš, J.; Zicha, J. Pathogenetic Mechanisms of Neurogenic Pulmonary Edema. J. Neurotrauma 2015, 32, 1135–1145. [Google Scholar] [CrossRef]
- West, J.B.; Mathieu-Costello, O. Stress failure of pulmonary capillaries in the intensive care setting. Schweiz. Med. Wochenschr. 1992, 122, 751–757. [Google Scholar]
- Bachofen, H.; Schürch, S.; Michel, R.P.; Weibel, E.R. Experimental hydrostatic pulmonary edema in rabbit lungs. Morphology. Am. Rev. Respir. Dis. 1993, 147, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Yabumoto, M.; Kuriyama, T.; Iwamoto, M.; Kinoshita, T. Neurogenic pulmonary edema associated with ruptured intracranial aneurysm: Case report. Neurosurgery 1986, 19, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Maire, F.W.; Patton, H.D. Role of the splanchnic nerve and the adrenal medulla in the genesis of preoptic pulmonary edema. Am. J. Physiol. 1956, 184, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Grauer, S.E.; Peterson, B.T.; Hyde, R.W.; Schwartz, S.I. Effect of autotransplantation of a lung on development of neurogenic pulmonary edema. Surg. Forum 1978, 29, 199–201. [Google Scholar] [PubMed]
- Avlonitis, V.S.; Wigfield, C.H.; Kirby, J.A.; Dark, J.H. The hemodynamic mechanisms of lung injury and systemic inflammatory response following brain death in the transplant donor. Am. J. Transplant. 2005, 5, 684–693. [Google Scholar] [CrossRef]
- Matsuyama, T.; Okuchi, K.; Nishiguchi, T.; Seki, T.; Murao, Y. Neurogenic pulmonary edema caused by a medulla oblongata lesion after head trauma. J. Trauma 2007, 63, 700–702. [Google Scholar] [CrossRef]
- Busl, K.M.; Bleck, T.P. Neurogenic Pulmonary Edema. Crit. Care Med. 2015, 43, 1710–1715. [Google Scholar] [CrossRef]
- Weir, B.K. Pulmonary edema following fatal aneurysm rupture. J. Neurosurg. 1978, 49, 502–507. [Google Scholar] [CrossRef]
- Legrand, M.; Sonneville, R. Understanding the renal response to brain injury. Intensive Care Med. 2019, 45, 1112–1115. [Google Scholar] [CrossRef]
- Adrogué, H.J.; Madias, N.E. Hyponatremia. N. Engl. J. Med. 2000, 25, 1581–1589. [Google Scholar] [CrossRef]
- Tisdall, M.; Crocker, M.; Watkiss, J.; Smith, M. Disturbances of sodium in critically ill adult neurologic patients: A clinical review. J. Neurosurg. Anesthesiol. 2006, 18, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, M.; Thompson, C.J. The syndrome of inappropriate antidiuresis (SIAD). Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Sterns, R.H. Disorders of plasma sodium. N. Engl. J. Med. 2015, 372, 1269. [Google Scholar] [CrossRef] [PubMed]
- Berendes, E.; Walter, M.; Cullen, P.; Prien, T.; Van Aken, H.; Horsthemke, J.; Schulte, M.; von Wild, K.; Scherer, R. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997, 349, 245–249. [Google Scholar] [CrossRef]
- Sterns, R.H.; Silver, S.M. Cerebral salt wasting versus SIADH: What difference? J. Am. Soc. Nephrol. 2008, 19, 194–196. [Google Scholar] [CrossRef]
- Vedantam, A.; Robertson, C.S.; Gopinath, S.P. Morbidity and mortality associated with hypernatremia in patients with severe traumatic brain injury. Neurosurg. Focus 2017, 43, 2. [Google Scholar] [CrossRef]
- Bistritzer, T.; Theodor, R.; Inbar, D.; Cohen, B.E.; Sack, J. Anterior hypopituitarism due to fracture of the sella turcica. Am. J. Dis. Child. 1981, 135, 966–968. [Google Scholar] [CrossRef]
- Sav, A.; Rotondo, F.; Syro, L.V.; Serna, C.A.; Kovac, K. Pituitary pathology in traumatic brain injury: A review. Pituitary 2019, 22, 201–211. [Google Scholar] [CrossRef]
- Seckl, J.R.; Dunger, D.B.; Lightman, S.L. Neurohypophyseal peptide function during early postoperative diabetes insipidus. Brain 1987, 110, 737–746. [Google Scholar] [CrossRef]
- Boughey, J.C.; Yost, M.J.; Bynoe, R.P. Diabetes insipidus in the head-injured patient. Am. Surg. 2004, 70, 500–503. [Google Scholar]
- Hannon, M.J.; Crowley, R.K.; Behan, L.A.; O’Sullivan, E.P.; O’Brien, M.M.; Sherlock, M.; Rawluk, D.; O’Dwyer, R.; Tormey, W.; Thompson, C.J. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J. Clin. Endocrinol. Metab. 2013, 98, 3229–3237. [Google Scholar] [CrossRef] [PubMed]
- Karali, V.; Massa, E.; Vassiliadou, G.; Chouris, I.; Rodin, I.; Bitzani, M. Evaluation of development of diabetes insipidus in the early phase following traumatic brain injury in critically ill patients. Crit. Care 2008, 12, 51–52. [Google Scholar] [CrossRef]
- Udy, A.; Boots, R.; Senthuran, S.; Stuart, J.; Deans, R.; Lassig-Smith, M.; Lipman, J. Augmented creatinine clearance in traumatic brain injury. Anesth. Analg. 2010, 111, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Udy, A.A.; Jarrett, P.; Lassig-Smith, M.; Stuart, J.; Starr, T.; Dunlop, R.; Deans, R.; Roberts, J.A.; Senthuran, S.; Boots, R.; et al. Augmented Renal Clearance in Traumatic Brain Injury: A Single-Center Observational Study of Atrial Natriuretic Peptide, Cardiac Output, and Creatinine Clearance. J. Neurotrauma 2017, 34, 137–144. [Google Scholar] [CrossRef]
- Khalid, F.; Yang, G.L.; McGuire, J.L.; Robson, M.J.; Foreman, B.; Ngwenya, L.B.; Lorenz, J.N. Autonomic dysfunction following traumatic brain injury: Translational insights. Neurosurg. Focus 2019, 47, 8. [Google Scholar] [CrossRef]
- Park, C.Y.; Choi, H.Y.; You, N.K.; Roh, T.H.; Seo, S.J.; Kim, S.H. Continuous Renal Replacement Therapy for Acute Renal Failure in Patients with Traumatic Brain Injury. Korean J. Neurotrauma 2016, 12, 89–93. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; Li, Z.; Li, M.; Han, Y.; Wang, L.; Zhang, Z.; Yu, C.; Zhang, F.; Song, L.; et al. Organ- and cell-specific immune responses are associated with the outcomes of intracerebral hemorrhage. FASEB J. 2018, 32, 220–229. [Google Scholar] [CrossRef]
- Busl, K.M. Nosocomial Infections in the Neurointensive Care Unit. Neurosurg. Clin. N. Am. 2018, 29, 299–314. [Google Scholar] [CrossRef]
- Esnault, P.; Nguyen, C.; Bordes, J.; D’Aranda, E.; Montcriol, A.; Contargyris, C.; Cotte, J.; Goutorbe, P.; Joubert, C.; Dagain, A.; et al. Early-Onset Ventilator-Associated Pneumonia in Patients with Severe Traumatic Brain Injury: Incidence, Risk Factors, and Consequences in Cerebral Oxygenation and Outcome. Neurocrit. Care 2017, 27, 187–198. [Google Scholar] [CrossRef]
- Klein, R.L.; Wilson, S.P.; Dzielak, D.J.; Yang, W.H.; Viveros, O.H. Opioid peptides and noradrenaline co-exist in large dense-cored vesicles from sympathetic nerve. Neuroscience 1982, 7, 2255–2261. [Google Scholar] [CrossRef]
- Sahota, P.; Vahidy, F.; Nguyen, C.; Bui, T.T.; Yang, B.; Parsha, K.; Garrett, J.; Bambhroliya, A.; Barreto, A.; Grotta, J.C.; et al. Changes in spleen size in patients with acute ischemic stroke: A pilot observational study. Int. J. Stroke 2013, 8, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Vahidy, F.S.; Parsha, K.N.; Rahbar, M.H.; Lee, M.; Bui, T.T.; Nguyen, C.; Barreto, A.D.; Bambhroliya, A.B.; Sahota, P.; Yang, B.; et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2016, 36, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Jacome, T.; Tatum, D. Systemic Inflammatory Response Syndrome (SIRS) Score Independently Predicts Poor Outcome in Isolated Traumatic Brain Injury. Neurocrit. Care 2018, 28, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Saand, A.R.; Yu, F.; Chen, J.; Chou, S.H. Systemic inflammation in hemorrhagic strokes—A novel neurological sign and therapeutic target? J. Cereb. Blood Flow Metab. 2019, 39, 959–988. [Google Scholar] [CrossRef] [PubMed]
- Brea, D.; Agulla, J.; Rodríguez-Yáñez, M.; Barral, D.; Ramos-Cabrer, P.; Campos, F.; Almeida, A.; Dávalos, A.; Castillo, J. Regulatory T cells modulate inflammation and reduce infarct volume in experimental brain ischaemia. J. Cell. Mol. Med. 2014, 18, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Subramanian, S.; Parker, S.M.; Wang, C.; Afentoulis, M.E.; Lewis, A.; Vandenbark, A.A.; Hurn, P.D. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J. Immunol. 2006, 176, 6523–6531. [Google Scholar] [CrossRef]
- Macrez, R.; Ali, C.; Toutirais, O.; Le Mauff, B.; Defer, G.; Dirnagl, U.; Vivien, D. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol. 2011, 10, 471–480. [Google Scholar] [CrossRef]
- Schorr, E.C.; Arnason, B.G. Interactions between the sympathetic nervous system and the immune system. Brain Behav. Immun. 1999, 13, 271–278. [Google Scholar] [CrossRef][Green Version]
- McCulloch, L.; Smith, C.J.; McColl, B.W. Adrenergic-mediated loss of splenic marginal zone B cells contributes to infection susceptibility after stroke. Nat. Commun. 2017, 19, 150–151. [Google Scholar]
- Madinier, A.; Bertrand, N.; Mossiat, C.; Prigent-Tessier, A.; Beley, A.; Marie, C.; Garnier, P. Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS ONE 2009, 4, e8101. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Kalil, A.; Shea, C.; Becker, K.J. Lymphocytes: Potential mediators of postischemic injury and neuroprotection. Stroke 2007, 38, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Peura, D.A.; Johnson, L.F. Cimetidine for prevention and treat- ment of gastroduodenal mucosal lesions in patients in an intensive care unit. Ann. Intern. Med. 1985, 103, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.F.; McAlhand, J.C.; Pruitt, B.A., Jr. Acute gastroduodenal disease after thermal injury. N. Engl. J. Med. 1974, 291, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Patejdl, R.; Kästner, M.; Kolbaske, S.; Wittstock, M. Clinical nutrition and gastrointestinal dysfunction in critically ill stroke patients. Neurol. Res. 2017, 39, 959–964. [Google Scholar] [CrossRef]
- Lewis, E.A. Gastroduodenal ulceration and haemorrhage of neurogenic origin. Br. J. Surg. 1973, 60, 279–283. [Google Scholar] [CrossRef]
- Kemp, W.J.; Bashir, A.; Dababneh, H.; Cohen-Gadol, A.A. Cushing’s ulcer: Further reflections. Asian J. Neurosurg. 2015, 10, 87–94. [Google Scholar]
- Cheung, L.Y. Thomas G Orr Memorial Lecture. Pathogenesis, prophylaxis, and treatment of stress gastritis. Am. J. Surg. 1988, 156, 437–440. [Google Scholar] [CrossRef]
- Sesler, J.M. Stress-related mucosal disease in the intensive care unit: An update on prophylaxis. AACN Adv. Crit. Care 2007, 18, 119–126. [Google Scholar] [CrossRef]
- Sadaka, F.; Trottier, S.; Smith, T.; VanSlette, J.; Kasal, J.; Palagiri, A. Proton Pump Inhibitors Versus Histamine 2 Receptor Antagonists for Stress Ulcer Prophylaxis. Crit. Care Med. 2013, 41, A224. [Google Scholar] [CrossRef]
- Lilly, C.M.; Aljawadi, M.; Badawi, O.; Onukwugha, E.; Tom, S.E.; Magder, L.S.; Harris, I. Comparative Effectiveness of Proton Pump Inhibitors vs Histamine Type 2 Receptor Blockers for Preventing Clinically Important Gastrointestinal Bleeding During Intensive Care: A Population-Based Study. Chest 2018, 154, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Krzych, Ł.J.; Łój, P.; Nowak, T.; Kazura, W.; Knapik, P. Short-term proton pump inhibitor treatment may cause hypomagnesaemia in critically ill patients—A pilot study. Acta Biochim. Pol. 2017, 64, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.F.; Bruno, J.J.; Buckley, M.S.; Cook, D.J. Stress Ulcer Prophylaxis. Crit. Care Med. 2016, 44, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Krag, M.; Perner, A.; Wetterslev, J.; Wise, M.P.; Hylander Møller, M. Stress ulcer prophylaxis versus placebo or no prophylaxis in critically ill patients. A systematic review of randomised clinical trials with meta-analysis and trial sequential analysis. Intensive Care Med. 2014, 40, 11–22. [Google Scholar] [CrossRef]
- El-Kersh, K.; Jalil, B.; McClave, S.A.; Cavallazzi, R.; Guardiola, J.; Guilkey, K.; Persaud, A.K.; Furmanek, S.P.; Guinn, B.E.; Wiemken, T.L.; et al. Enteral nutrition as stress ulcer prophylaxis in critically ill patients: A randomized controlled exploratory study. J. Crit. Care 2018, 43, 108–113. [Google Scholar] [CrossRef]
- Hurt, R.T.; Frazier, T.H.; McClave, S.A.; Crittenden, N.E.; Kulisek, C.; Saad, M.; Franklin, G.A. Stress prophylaxis in intensive care unit patients and the role of enteral nutrition. J. Parenter. Enter. Nutr. 2012, 36, 721–731. [Google Scholar] [CrossRef]
- Yu, K.; Zheng, X.; Wang, G.; Liu, M.; Li, Y.; Yu, P.; Yang, M.; Guo, N.; Ma, X.; Bu, Y.; et al. Immunonutrition vs Standard Nutrition for Cancer Patients: A Systematic Review and Meta-Analysis (Part 1). J. Parenter. Enter. Nutr. 2020, 44, 742–767. [Google Scholar] [CrossRef]
- Rai, V.R.H.; Phang, L.F.; Sia, S.F.; Amir, A.; Veerakumaran, J.S.; Kassim, M.K.A.; Othman, R.; Tah, P.C.; Loh, P.S.; Jailani, M.I.O.; et al. Effects of immunonutrition on biomarkers in traumatic brain injury patients in Malaysia: A prospective randomized controlled trial. BMC Anesthesiol. 2017, 17, 81. [Google Scholar] [CrossRef]
- Arya, A.K.; Hu, B. Brain-gut axis after stroke. Brain Circ. 2018, 4, 165–173. [Google Scholar] [CrossRef]
- De Jong, P.R.; González-Navajas, J.M.; Jansen, N.J. The digestive tract as the origin of systemic inflammation. Crit. Care 2016, 20, 279. [Google Scholar] [CrossRef]
- Yoseph, B.P.; Klingensmith, N.J.; Liang, Z.; Breed, E.R.; Burd, E.M.; Mittal, R.; Dominguez, J.A.; Petrie, B.; Ford, M.L.; Coopersmith, C.M. Mechanisms of Intestinal Barrier Dysfunction in Sepsis. Shock 2016, 46, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wiórek, A.; Jaworski, T.; Krzych, Ł.J. Hyperosmolar Treatment for Patients at Risk for Increased Intracranial Pressure: A Single-Center Cohort Study. Int. J. Environ. Res. Public Health 2020, 17, 4573. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Hormone/-s | Methods of Assessment | Signs and Symptoms in the Acute Phase | Signs and Symptoms in the Chronic Phase | Need of Replacement Therapy | ||
|---|---|---|---|---|---|---|
| Antidiuretic Hormone | ||||||
| SIADH | hyponatremia (<135 mmol/L) urine osmolality > 100 mOsm/kg | Oliguria | As in the chronic phase | - | ||
| Central DI | hypernatremia (>145 mmol/L) urine osmolality < 250 mOsm/kg H2O low serum vasopressin levels | Polyuria Polydypsia | As in the chronic phase | If central DI is present—vasopressin replacement therapy with desmopressin | ||
| Growth hormone (GH) | Levels of GH and insulin-like growth factor 1 (IGF-1), GH response after growth hormone-releasing hormone (GHRH) | Not clinically evident | Poor quality of life, low mood, fatigue, cognitive dysfunction, reduction in muscle mass, increased body fat, decreased exercise capacity, increased lipid levels, reduced body mineral density, reduced LV mass, impaired LVEF | May partially reverse cognitive dysfunction after TBI. | ||
| Adrenal hormones (mineralocorticoid and glucocorticoid) | Serum cortisol concentration (<15–18 μg/dL/413–497 nmol/L), ACTH stimulation test (a change ≥9 μg/dL/248 nmol/L—adequate response) | Greater requirements for vasoactive therapy, hypoglycemia, hyponatremia, relative or absolute hyperkalaemia, rapidly progressive hypotension with hyperdynamic cardiovascular response with low SVR | Recurrent infections, fatigue, weight loss, nauseas, vomiting, hypoglycemia (mostly fasting), anorexia, myalgia Adrenal crisis: hypotension, hyponatremia, hypercalcemia, hyperkalaemia, syncope | Hormonal replacement therapy should be provided if hypoadrenalism have been confirmed. Hydrocortisone should be administered in all cases of adrenal failure, however mineralocorticoid supplemental is recommended if primary failure occurs. | ||
| TSH, T3, T4 | Level of total serum T3 and T4, TSH concentration | May resemble symptoms of already acutely ill patients: impaired consciousness, myocardial dysfunction, hypothermia, neuropathy, muscle weakness, skin atrophy | Dry skin, excessive weight, bradycardia, systemic hypertension, fatigue, constipation, cold intolerance, muscle aches, vocal changes, prolonged ankle-jerk reflex time, hyponatremia | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachfalska, N.; Putowski, Z.; Krzych, Ł.J. Distant Organ Damage in Acute Brain Injury. Brain Sci. 2020, 10, 1019. https://doi.org/10.3390/brainsci10121019
Rachfalska N, Putowski Z, Krzych ŁJ. Distant Organ Damage in Acute Brain Injury. Brain Sciences. 2020; 10(12):1019. https://doi.org/10.3390/brainsci10121019
Chicago/Turabian StyleRachfalska, Natalia, Zbigniew Putowski, and Łukasz J. Krzych. 2020. "Distant Organ Damage in Acute Brain Injury" Brain Sciences 10, no. 12: 1019. https://doi.org/10.3390/brainsci10121019
APA StyleRachfalska, N., Putowski, Z., & Krzych, Ł. J. (2020). Distant Organ Damage in Acute Brain Injury. Brain Sciences, 10(12), 1019. https://doi.org/10.3390/brainsci10121019