Synthesis, Tyrosinase Inhibiting Activity and Molecular Docking of Fluorinated Pyrazole Aldehydes as Phosphodiesterase Inhibitors

 , ,
, ,  ,
,  ,
, 
Abstract
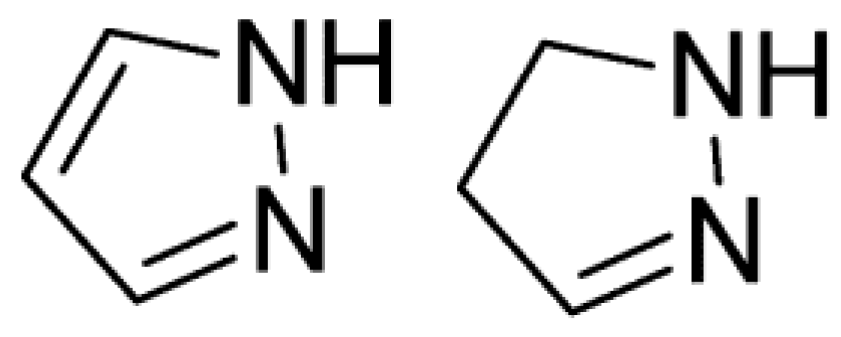
1. Introduction
2. Materials and Methods
2.1. General
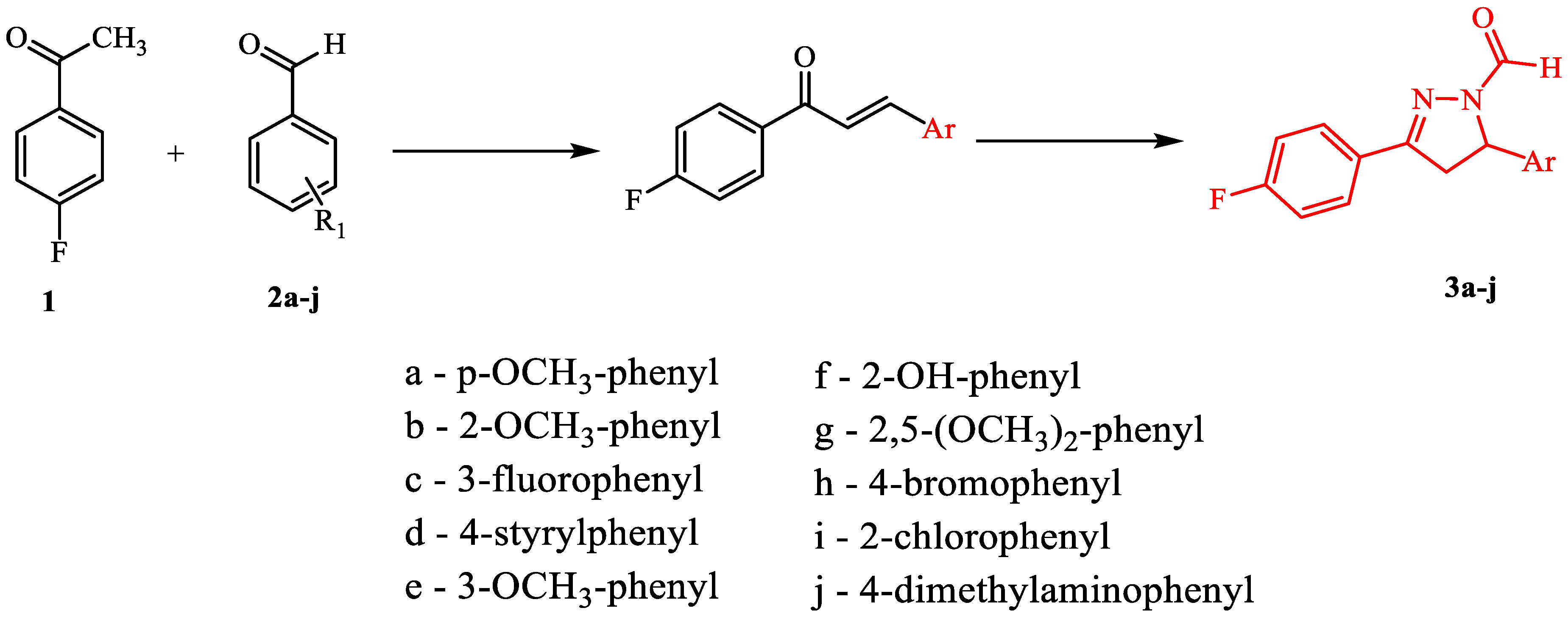
2.2. General Procedure for Synthesis of Fluorinated Pyrazoles (3a–j)
2.3. Antibacterial Susceptibility Testing
2.4. Tyrosinase Inhibiting Activity
2.5. Docking Studies
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alam, J.; Alam, O.; Alam, P.; Naim, M.J. A Review on pyrazole chemical entity and biological activity. Int. J. Pharm. Sci. Res. 2015, 6, 1433–1442. [Google Scholar]
- Alex, J.M.; Kumar, R. 4,5-Dihydro-1H-pyrazole: An indispensable scaffold. J. Enzyme Inhib. Med. Chem. 2014, 29, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Shingare, R.M.; Patil, Y.S.; Gadekar, S.; Sangshetti, J.N.; Madje, B.R. Synthesis and antibacterial screening of novel 1,3,5-triaryl-4,5-dihydro-1H-pyrazole derivatives. Morocc. J. Chem. 2017, 5, 177–185. [Google Scholar]
- Zhao, M.-Y.; Yin, Y.; Yu, X.-W.; Sangani, C.B.; Wang, S.-F.; Lu, A.-M.; Yang, L.-F.; Lv, P.-C.; Jiang, M.-G.; Zhu, H.-L. Synthesis, biological evaluation and 3D-QSAR study of novel 4,5-dihydro-1H-pyrazole thiazole derivatives as BRAFV600E inhibitors. Bioorg. Med. Chem. 2015, 23, 46–54. [Google Scholar] [PubMed]
- Zhou, Z.; Zhuo, J.; Yan, S.; Ma, L. Design and synthesis of 3,5-diaryl-4,5-dihydro-1H-pyrazoles as new tyrosinase inhibitors. Bioorg. Med. Chem. 2013, 21, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Strunecká, A.; Patočka, J.; Connett, P. Fluorine in medicine. J. Appl. Biomed. 2004, 2, 141–150. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Banday, A.H.; Mir, B.P.; Lone, I.H.; Suri, K.A.; Kumar, H.M.S. Studies on novel D-ring substituted steroidal pyrazolines as potential anticancer agents. Steroids 2010, 75, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, F.F.; Girgis, A.S. Facile synthesis of bis(4,5-dihydro-1H-pyrazole-1-carboxamides) and their thio-analogues of potential PGE(2) inhibitory properties. Eur. J. Med. Chem. 2009, 44, 2172–2177. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Adsul, L.K.; Chavan, H.V.; Jalde, S.S.; Shringare, S.N.; Shaikh, R.; Meshram, R.J.; Gacche, R.N.; Masand, V. Synthesis, biological evaluation, and docking studies of 3-(substituted)-aryl-5-(9-methyl-3-carbazole)-1H-2-pyrazolines as potent anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 5839–5844. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, Z.; Kandilci, H.B.; Gümüşel, B.; Caliş, U.; Bilgin, A.A. Synthesis and studies on antidepressant and anticonvulsant activities of some 3-(2-furyl)-pyrazoline derivatives. Eur. J. Med. Chem. 2007, 42, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Sachchar, S.P.; Singh, A.K. Synthesis of some new fluorinated heteroaryl pyrazolines and isooxazolines as potential biocidal agents. J. Indian Chem. Soc. 1986, 62, 142–146. [Google Scholar] [CrossRef]
- Camacho, M.E.; León, J.; Entrena, A.; Velasco, G.; Carrión, M.D.; Escames, G.; Vivó, A.; Acuña-Castroviejo, D.; Gallo, M.A.; Espinosa, A. 4,5-Dihydro-1H-pyrazole derivatives with inhibitory nNOS activity in rat brain: Synthesis and structure−Activity relationships. J. Med. Chem. 2004, 47, 5641–5650. [Google Scholar] [CrossRef] [PubMed]
- Dabholkar, V.; Ansari, F. Synthesis and characterization of selected fused isoxazole and pyrazole derivatives and their antimicrobial activity. J. Serb. Chem. Soc. 2009, 74, 1219–1228. [Google Scholar] [CrossRef]
- Gressler, V.; Moura, S.; Flores, A.F.C.; Flores, D.C.; Colepicolo, P.; Pinto, E. Antioxidant and antimicrobial properties of 2-(4,5-dihydro-1H-pyrazol-1-yl)-pyrimidine and 1-carboxamidino-1H-pyrazole derivatives. J. Braz. Chem. Soc. 2010, 21, 1477–1483. [Google Scholar] [CrossRef]
- Liu, J.-J.; Sun, J.; Fang, Y.-B.; Yang, Y.-A.; Jiao, R.-H.; Zhu, H.-L. Synthesis, and antibacterial activity of novel 4,5-dihydro-1H-pyrazole derivatives as DNA gyrase inhibitors. Org. Biomol. Chem. 2014, 12, 998–1008. [Google Scholar] [CrossRef]
- Rostom, S.A.F.; Badr, M.H.; Abd El Razik, H.A.; Ashour, H.M.A.; Abdel Wahab, A.E. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Arch. Pharm. 2011, 344, 572–587. [Google Scholar] [CrossRef]
- Molnar, M.; Kovač, T.; Strelec, I. Umbelliferone-thiazolidinedione hybrids as potent mushroom tyrosinase inhibitors. Int. J. Pharm. Res. Allied Sci. 2016, 5, 305–310. [Google Scholar]
- Poroikov, V.V.; Filimonov, D.A.; Ihlenfeldt, W.-D.; Gloriozova, T.A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M.C. PASS biological activity spectrum predictions in the enhanced open NCI database browser. J. Chem. Inf. Comput. Sci. 2003, 43, 228–236. [Google Scholar] [CrossRef]
- Hocquet, A.; Langgård, M. An evaluation of the MM+ force field. J. Mol. Med. 1998, 4, 94–112. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Ren, J.; He, Y.; Chen, W.; Chen, T.; Wang, G.; Wang, Z.; Xu, Z.; Luo, X.; Zhu, W.; Jiang, H.; et al. Thermodynamic and structural characterization of halogen bonding in protein–ligand interactions: A case study of PDE5 and its inhibitors. J. Med. Chem. 2014, 57, 3588–3593. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Perkins, H.R. Microbial Cell Walls and Membranes; Springer: Dordrecht, The Netherlands, 1980; ISBN 978-94-011-6016-2. [Google Scholar]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Schellack, N.; Agoro, A. A review of phosphodiesterase type 5 inhibitors. S. Afr. Fam. Pract. 2014, 56, 96–101. [Google Scholar] [CrossRef]
- Wang, X.-H.; Wang, X.-K.; Liang, Y.-J.; Shi, Z.; Zhang, J.-Y.; Chen, L.-M.; Fu, L.-W. A cell-based screen for anticancer activity of 13 pyrazolone derivatives. Chin. J. Cancer 2010, 29, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.-J.; Hwang, K.Y.; Jeon, Y.H.; Lee, J.I.; Heo, Y.-S.; Kim, J.H.; Moon, J.; Yoon, J.M.; Hyun, Y.-L.; Kim, E.; et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature 2003, 425, 98–102. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
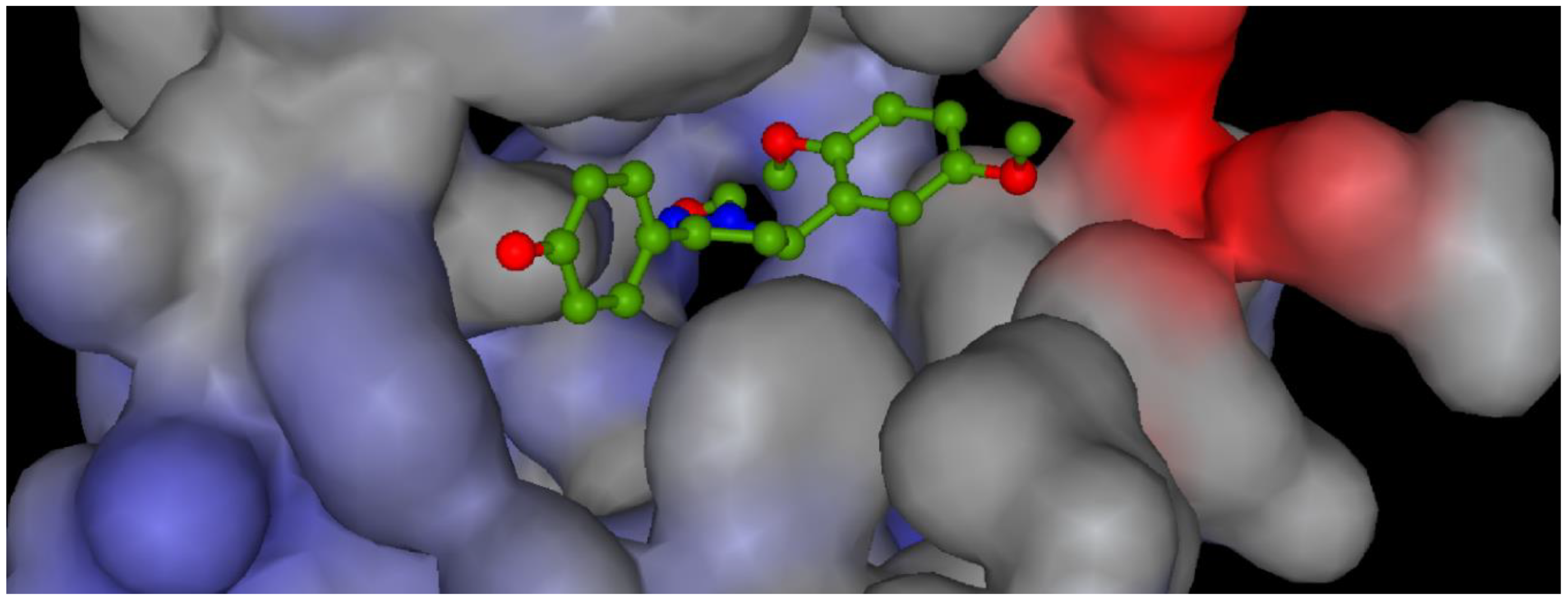{kind=link}
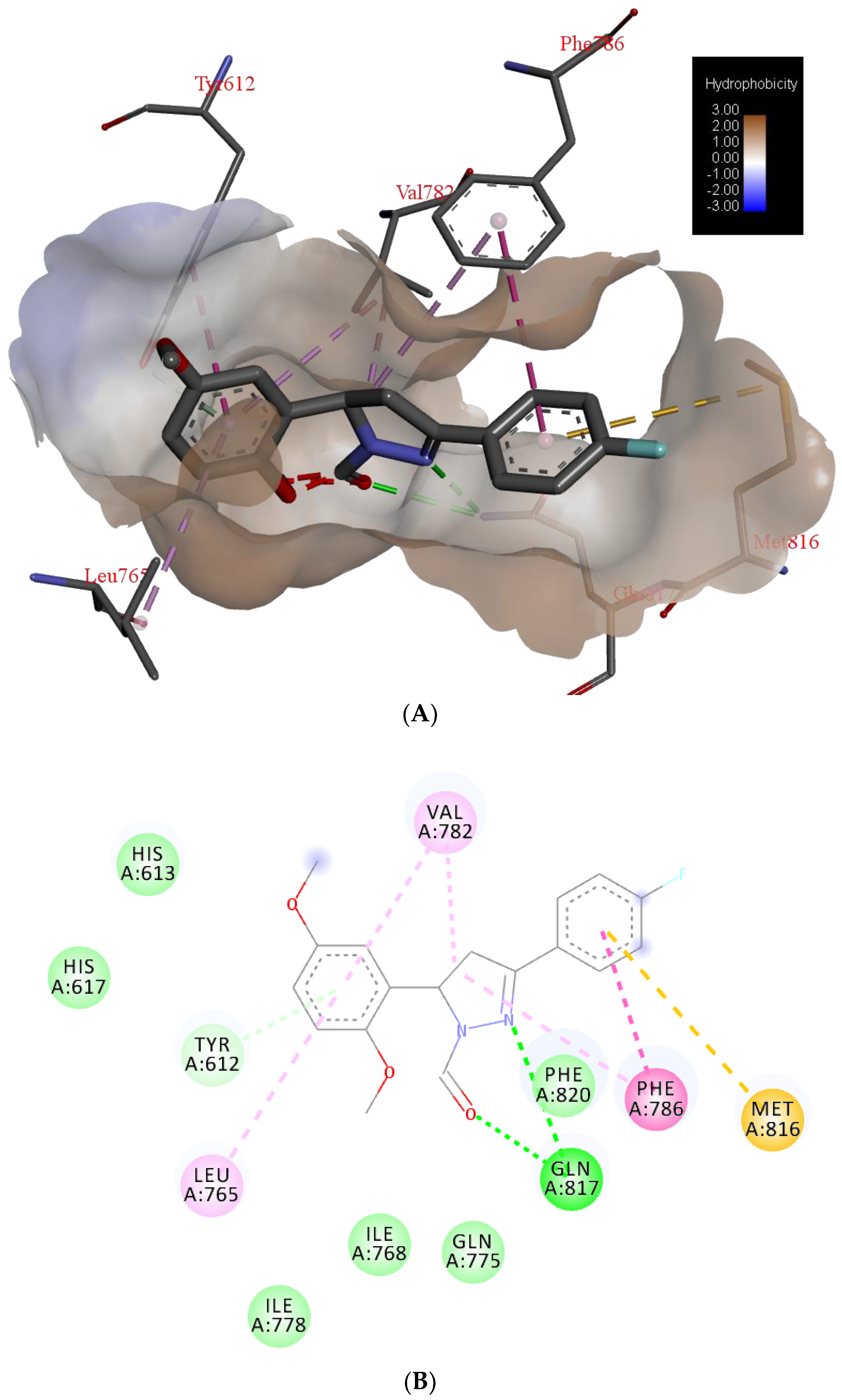{kind=link}
| Compound | Minimum Inhibitory Concentration (μg mL−1) | |||
|---|---|---|---|---|
| E. coli | P. aeruginosa | B. subtilis | S. aureus | |
| 3a | 62.5 | 62.5 | 62.5 | 62.5 |
| 3b | 62.5 | 62.5 | 62.5 | 62.5 |
| 3c | 62.5 | 62.5 | 62.5 | 62.5 |
| 3d | 62.5 | 62.5 | 62.5 | 62.5 |
| 3e | 62.5 | 62.5 | 62.5 | 62.5 |
| 3f | 62.5 | 62.5 | 62.5 | 62.5 |
| 3g | 62.5 | 62.5 | 125 | 125 |
| 3h | 62.5 | 62.5 | 250 | 250 |
| 3i | 62.5 | 62.5 | 250 | 250 |
| 3j | 62.5 | 62.5 | 250 | 250 |
| amikacin | 1.95 | 0.49 | 0.24 | 1.95 |
| Compound | Monophenolase Inhibition Rate (%) | Diphenolase Inhibition Rate (%) |
|---|---|---|
| 3a | 10.84 ± 1.20 | 21.08 ± 1.86 |
| 3b | 5.22 ± 1.84 | 15.98 ± 3.25 |
| 3c | 0.21 ± 1.39 | 10.11 ± 3.92 |
| 3d | 28.80 ± 4.10 | 22.81 ± 0.33 |
| 3e | 21.74 ± 2.82 | 16.55 ± 0.29 |
| 3f | 20.11 ± 1.63 | 13.67 ± 0.50 |
| 3g | 25.54 ± 2.49 | 14.34 ± 0.88 |
| 3h | 24.46 ± 3.77 | 20.79 ± 0.17 |
| 3i | 32.07 ± 3.39 | 15.11 ± 0.50 |
| 3j | 25.54 ± 4.10 | 18.38 ± 1.01 |
| Kojic acid | 100 ± 0.00 | 88.45 ± 0.83 |
| Compound | Pa * | Pi |
|---|---|---|
| 3a | 0.651 | 0.004 |
| 3b | 0.584 | 0.004 |
| 3c | 0.616 | 0.004 |
| 3d | 0.516 | 0.004 |
| 3e | 0.637 | 0.004 |
| 3f | 0.396 | 0.005 |
| 3g | 0.611 | 0.004 |
| 3h | 0.469 | 0.004 |
| 3i | 0.508 | 0.004 |
| 3j | 0.460 | 0.005 |
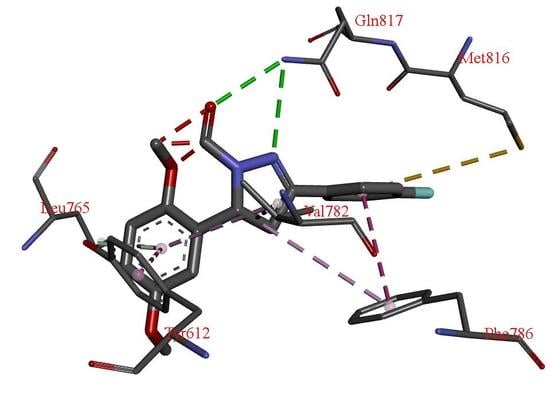
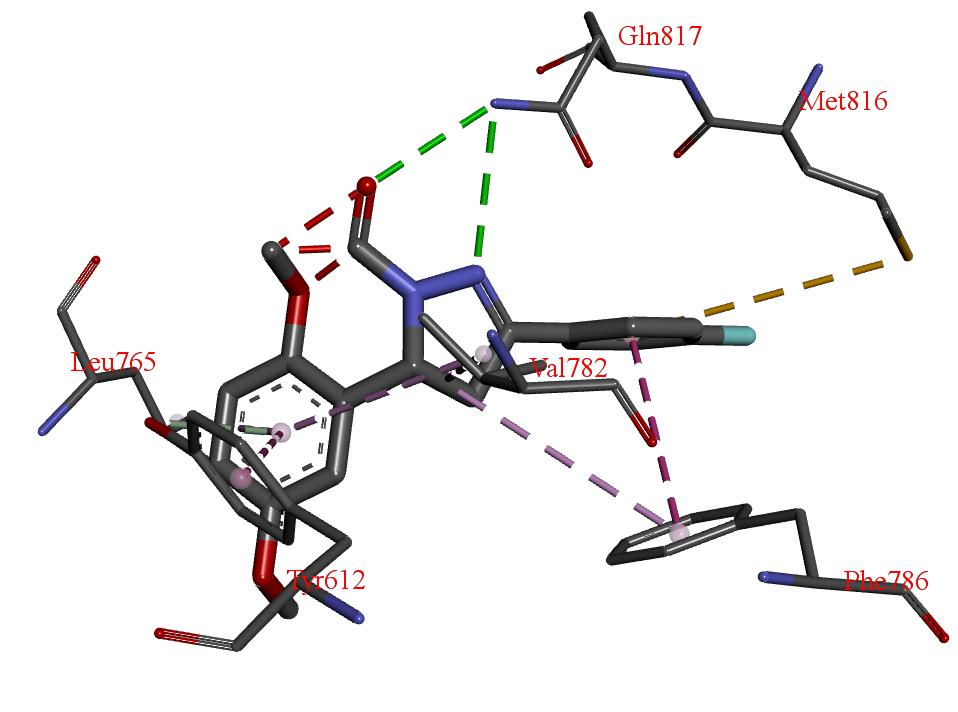
| Comp. | Pose | Total Energy/kcal mol−1 | Van Der Waals Interaction | H Bond | Elec |
|---|---|---|---|---|---|
| 3g | 1 | −105.62 | −95.34 | −10.27 | 0 |
| 3f | 1 | −98.66 | −75.26 | −23.40 | 0 |
| 3b | 0 | −98.44 | −90.79 | −7.66 | 0 |
| 3d | 0 | −96.70 | −82.39 | −14.31 | 0 |
| 3i | 0 | −93.07 | −84.42 | −8.65 | 0 |
| 3e | 1 | −89.18 | −84.61 | −4.58 | 0 |
| 3a | 0 | −88.90 | −66.30 | −22.60 | 0 |
| 3c | 0 | −88.78 | −81.78 | −7.00 | 0 |
| 3j | 0 | −88.49 | −86.64 | −1.85 | 0 |
| 3h | 0 | −88.09 | −70.57 | −17.52 | 0 |
| H Bond | Energy | Van Der Waals Interaction | Energy |
|---|---|---|---|
| S-Gln817 | −6.81 | S-Phe820 | −18.51 |
| S-His613 | −3.41 | S-Tyr612 | −10.33 |
| S-Val782 | −8.70 | ||
| S-Phe786 | −7.90 | ||
| M-Leu765 | −4.20 | ||
| S-Leu765 | −4.04 | ||
| S-Met816 | −4.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rastija, V.; Brahmbhatt, H.; Molnar, M.; Lončarić, M.; Strelec, I.; Komar, M.; Pavić, V. Synthesis, Tyrosinase Inhibiting Activity and Molecular Docking of Fluorinated Pyrazole Aldehydes as Phosphodiesterase Inhibitors. Appl. Sci. 2019, 9, 1704. https://doi.org/10.3390/app9081704
Rastija V, Brahmbhatt H, Molnar M, Lončarić M, Strelec I, Komar M, Pavić V. Synthesis, Tyrosinase Inhibiting Activity and Molecular Docking of Fluorinated Pyrazole Aldehydes as Phosphodiesterase Inhibitors. Applied Sciences. 2019; 9(8):1704. https://doi.org/10.3390/app9081704
Chicago/Turabian StyleRastija, Vesna, Harshad Brahmbhatt, Maja Molnar, Melita Lončarić, Ivica Strelec, Mario Komar, and Valentina Pavić. 2019. "Synthesis, Tyrosinase Inhibiting Activity and Molecular Docking of Fluorinated Pyrazole Aldehydes as Phosphodiesterase Inhibitors" Applied Sciences 9, no. 8: 1704. https://doi.org/10.3390/app9081704
APA StyleRastija, V., Brahmbhatt, H., Molnar, M., Lončarić, M., Strelec, I., Komar, M., & Pavić, V. (2019). Synthesis, Tyrosinase Inhibiting Activity and Molecular Docking of Fluorinated Pyrazole Aldehydes as Phosphodiesterase Inhibitors. Applied Sciences, 9(8), 1704. https://doi.org/10.3390/app9081704