The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges


 ,
,  ,
,  ,
, 
Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Synthesis of Amines 36, 37
2.1.2. General Procedure for Amides 44a,b; 45a,b; 50a,b
2.1.3. N-(3,7-Dimethyloctyl)adamantane-1-carboxamide 44a
2.1.4. N-((Z)-3,7-Dimethylocta-2,6-dien-1-yl)adamantane-1-carboxamide 44b
2.1.5. N-(3,7-Dimethyloctyl)adamantane-2-carboxamide 45a
2.1.6. N-((Z)-3,7-Dimethylocta-2,6-dien-1-yl)adamantane-2-carboxamide 45b
2.1.7. N-(Adamantan-1-yl)-3,7-dimethyloct-6-enamide 50a
2.1.8. N-(Adamantan-2-yl)-3,7-dimethyloct-6-enamide 50b
2.1.9. General Procedure for Thioamides 46a,b; 47a,b; 51
2.1.10. N-(3,7-Dimethyloctyl)adamantane-1-carbothioamide 46a
2.1.11. N-((Z)-3,7-Dimethylocta-2,6-dien-1-yl)adamantane-1-carbothioamide 46b
2.1.12. N-(3,7-Dimethyloctyl)adamantane-2-carbothioamide 47a
2.1.13. N-((Z)-3,7-Dimethylocta-2,6-dien-1-yl)adamantane-2-carbothioamide 47b
2.1.14. N-(Adamantan-2-yl)-3,7-dimethyloct-6-enethioamide 51
2.2. Modelling and Screening
2.3. Biological Study
2.3.1. TDP1 Activity
2.3.2. Cytotoxicity Assay
2.3.3. Binding Studies
3. Results and Discussion
3.1. Organic Synthesis
3.2. Inhibition Studies
3.3. Cytotoxicity
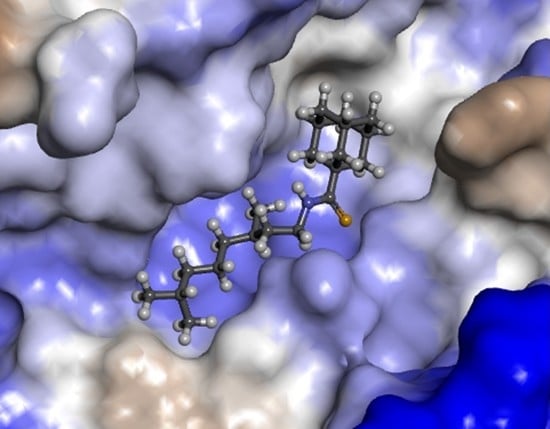
3.4. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Hevener, K.E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [PubMed]
- Cuya, S.M.; Bjornsti, M.-A.; van Waardenburg, R.C.A.M. DNA topoisomerase-targeting chemotherapeutics: what’s new? Cancer Chemother. Pharmacol. 2017, 80, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; Lavrik, O.I. AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett. 2011, 585, 683–686. [Google Scholar] [CrossRef]
- Pommier, Y.; Redon, C.; Rao, V.A.; Seiler, J.A.; Sordet, O.; Takemura, H.; Antony, S.; Meng, L.H.; Liao, Z.Y.; Kohlhagen, G. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. -Fundam. Mol. Mech. Mutagen. 2003, 532, 173–203. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, A.-K.; Lauridsen, K.L.; Samuel, E.B.; Proszek, J.; Knudsen, B.R.; Hager, H.; Stougaard, M. Correlation between topoisomerase I and tyrosyl-DNA phosphodiesterase 1 activities in non-small cell lung cancer tissue. Exp. Mol. Pathol. 2015, 99, 56–64. [Google Scholar] [CrossRef]
- Meisenberg, C.; Gilbert, D.C.; Chalmers, A.; Haley, V.; Gollins, S.; Ward, S.E.; El-Khamisy, S.F. Clinical and Cellular Roles for TDP1 and TOP1 in Modulating Colorectal Cancer Response to Irinotecan. Mol. Cancer Ther. 2015, 14, 575–585. [Google Scholar] [CrossRef]
- Perego, P.; Cossa, G.; Tinelli, S.; Corna, E.; Carenini, N.; Gatti, L.; De Cesare, M.; Ciusani, E.; Zunino, F.; Luison, E. Role of tyrosyl-DNA phosphodiesterase 1 and inter-players in regulation of tumor cell sensitivity to topoisomerase I inhibition. Biochem. Pharmacol. 2012, 83, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Dexheimer, T.S.; Takeda, S.; Pommier, Y. Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells. J. Biol. Chem. 2012, 287, 12848–12857. [Google Scholar] [CrossRef]
- Alagoz, M.; C. Gilbert, D.; El-Khamisy, S.; J. Chalmers, A. DNA Repair and Resistance to Topoisomerase I Inhibitors: Mechanisms, Biomarkers and Therapeutic Targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.N.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J. DNA-Damaging Anticancer Drugs—A Perspective for DNA Repair-Oriented Therapy. Curr. Med. Chem. 2017, 24, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Zakharova, O.D.; Rakhmanova, M.E.; Chepanova, A.A.; Dyrkheeva, N.S.; Lavrik, O.I.; Salakhutdinov, N.F. Usnic acid derivatives are effective inhibitors of tyrosyl-DNA phosphodiesterase 1. Russ. J. Biorgan. Chem. 2017, 43, 84–90. [Google Scholar] [CrossRef]
- Liao, Z.; Thibaut, L.; Jobson, A.; Pommier, Y. Inhibition of human Tyrosyl-DNA Phosphodiesterase (Tdp1) by aminoglycoside antibiotics and ribosome inhibitors. Mol. Pharmacol. 2006, 70, 366–372. [Google Scholar] [CrossRef]
- Komarova, A.O.; Drenichev, M.S.; Dyrkheeva, N.S.; Kulikova, I.V.; Oslovsky, V.E.; Zakharova, O.D.; Zakharenko, A.L.; Mikhailov, S.N.; Lavrik, O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enzyme Inhib. Med. Chem. 2018, 33, 1415–1429. [Google Scholar] [CrossRef]
- Nguyen, T.X.; Morrell, A.; Conda-Sheridan, M.; Marchand, C.; Agama, K.; Bermingam, A.; Stephen, A.G.; Chergui, A.; Naumova, A.; Fisher, R. Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)–Topoisomerase I (Top1) Inhibitors. J. Med. Chem. 2012, 55, 4457–4478. [Google Scholar] [CrossRef]
- Cushman, M.S.; Wang, P.; Pomier, Y.G.; Elsayed, M.S.A. Azaindenoisoquinoline Compounds and Uses Thereof. Patent Application WO 2018/118852 A1, 28 June 2018. [Google Scholar]
- Cushman, M.S.; Beck, D.E.; Pomier, Y.G. Aza-A-Ring Indenoisoquinoline Topoisomerase I Poisons. Patent Application WO2017160898A1, 21 September 2017. [Google Scholar]
- Beck, D.E.; Reddy, P.V.N.; Lv, W.; Abdelmalak, M.; Tender, G.S.; Lopez, S.; Agama, K.; Marchand, C.; Pommier, Y.; Cushman, M. Investigation of the Structure-Activity Relationships of Aza-A-Ring Indenoisoquinoline Topoisomerase I Poisons. J. Med. Chem. 2016, 59, 3840–3853. [Google Scholar] [CrossRef]
- Pommier, Y.; Thibaut, L.; Marchand, C. Tetracycline Compounds and Methods of Treatment. Patent Application WO 2007/112121 A2, 26 June 2001. [Google Scholar]
- Zhang, X.-R.; Wang, H.-W.; Tang, W.-L.; Zhang, Y.; Yang, H.; Hu, D.-X.; Ravji, A.; Marchand, C.; Kiselev, E.; Ofori-Atta, K. Discovery, Synthesis, and Evaluation of Oxynitidine Derivatives as Dual Inhibitors of DNA Topoisomerase IB (TOP1) and Tyrosyl-DNA Phosphodiesterase 1 (TDP1), and Potential Antitumor Agents. J. Med. Chem. 2018, 61, 9908–9930. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.S.; Gediya, L.K.; Stephen, A.G.; Weidlich, I.; Antony, S.; Marchand, C.; Interthal, H.; Nicklaus, M.; Fisher, R.J.; Njar, V.C. 4-Pregnen-21-ol-3,20-dione-21-(4-bromobenzenesufonate) (NSC 88915) and related novel steroid derivatives as tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors. J. Med. Chem. 2009, 52, 7122–7131. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Marchand, C.; Wang, Z.; Sirivolu, V.R. Thioxothiazolidinone Derivatives Useful as Inhibitors of Tdp1. WO2013/055771 A1, 18 April 2013. [Google Scholar]
- Sirivolu, V.R.; Vernekar, S.K.V.; Marchand, C.; Naumova, A.; Chergui, A.; Renaud, A.; Stephen, A.G.; Chen, F.; Sham, Y.Y.; Pommier, Y. 5-Arylidenethioxothiazolidinones as Inhibitors of Tyrosyl-DNA Phosphodiesterase I. J. Med. Chem. 2012, 55, 8671–8684. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.H.; Kumar, V.; Dexheimer, T.S.; Wedlich, I.; Nicklaus, M.C.; Pommier, Y.; Malhotra, S.V. Synthesis, anti-cancer screening and tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibition activity of novel piperidinyl sulfamides. Eur. J. Pharm. Sci. 2018, 111, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Biorgan. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Marchand, C.; Selvam, P.; Dexheimer, T.; Maddali, K. Fluoroquinolone Derivatives or Sulfonamide Moiety-Containing Compounds as Inhibitors of Tyrosyl-Dnaphosphodiesterase (TDP1). US8716295 (B2), 6 May 2014. [Google Scholar]
- Pommier, Y.; Marchand, C. Diamidine Inhibitors of TDP1. WO2007/126857 A1, 8 November 2007. [Google Scholar]
- Antony, S.; Marchand, C.; Stephen, A.G.; Thibaut, L.; Agama, K.K.; Fisher, R.J.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, O.; Luzina, O.; Zakharenko, A.; Sokolov, D.; Filimonov, A.; Dyrkheeva, N.; Chepanova, A.; Ilina, E.; Ilyina, A.; Klabenkova, K. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Biorgan. Med. Chem. 2018, 26, 4470–4480. [Google Scholar] [CrossRef]
- Dyrkheeva, N.; Luzina, O.; Filimonov, A.; Zakharova, O.; Ilina, E.; Zakharenko, A.; Kuprushkin, M.; Nilov, D.; Gushchina, I.; Švedas, V. Inhibitory Effect of New Semisynthetic Usnic Acid Derivatives on Human Tyrosyl-DNA Phosphodiesterase 1. Planta Med. 2019, 85, 103–111. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef]
- Bermingham, A.; Price, E.; Marchand, C.; Chergui, A.; Naumova, A.; Whitson, E.L.; Krumpe, L.R.H.; Goncharova, E.I.; Evans, J.R.; McKee, T.C. Identification of Natural Products That Inhibit the Catalytic Function of Human Tyrosyl-DNA Phosphodiesterase (TDP1). SLAS Discov. Adv. Life Sci. R D 2017, 22, 1093–1105. [Google Scholar] [CrossRef]
- Tian, L.-W.; Feng, Y.; Tran, T.D.; Shimizu, Y.; Pfeifer, T.; Vu, H.T.; Quinn, R.J. Achyrodimer F, a tyrosyl-DNA phosphodiesterase I inhibitor from an Australian fungus of the family Cortinariaceae. Biorgan. Med. Chem. Lett. 2017, 27, 4007–4010. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.-W.; Feng, Y.; Tran, T.D.; Shimizu, Y.; Pfeifer, T.; Forster, P.I.; Quinn, R.J. Tyrosyl-DNA Phosphodiesterase I Inhibitors from the Australian Plant Macropteranthes leichhardtii. J. Nat. Prod. 2015, 78, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Weidlich, I.E.; Dexheimer, T.; Marchand, C.; Antony, S.; Pommier, Y.; Nicklaus, M.C. Inhibitors of human tyrosyl-DNA phospodiesterase (hTdp1) developed by virtual screening using ligand-based pharmacophores. Biorgan. Med. Chem. 2010, 18, 182–189. [Google Scholar] [CrossRef]
- Salakhutdinov, N.F.; Volcho, K.P.; Yarovaya, O.I. Monoterpenes as a renewable source of biologically active compounds. Pure Appl. Chem. 2017, 89, 1105–1117. [Google Scholar] [CrossRef]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Biorgan. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef]
- Mozhaitsev, E.; Suslov, E.; Demidova, Y.; Korchagina, D.; Volcho, K.; Zakharenko, A.; Vasil’eva, I.; Kupryushkin, M.; Chepanova, A.; Ayine-Tora, D.M. The Development of Tyrosyl-DNA Phosphodyesterase 1 (TDP1) Inhibitors Based on the Amines Combining Aromatic/Heteroaromatic and Monoterpenoid Moieties. Lett. Drug Des. Discov. 2019, 16, 597–605. [Google Scholar] [CrossRef]
- Salomatina, O.; Popadyuk, I.; Zakharenko, A.; Zakharova, O.; Fadeev, D.; Komarova, N.; Reynisson, J.; Arabshahi, H.; Chand, R.; Volcho, K. Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Molecules 2018, 23, 679. [Google Scholar] [CrossRef]
- Li-Zhulanov, N.; Zakharenko, A.; Chepanova, A.; Patel, J.; Zafar, A.; Volcho, K.; Salakhutdinov, N.; Reynisson, J.; Leung, I.; Lavrik, O. A Novel Class of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors That Contains the Octahydro-2H-chromen-4-ol Scaffold. Molecules 2018, 23, 2468. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Ponomarev, K.U.; Suslov, E.V.; Korchagina, D.V.; Volcho, K.P.; Vasil’eva, I.A.; Salakhutdinov, N.F.; Lavrik, O.I. Inhibitory properties of nitrogen-containing adamantane derivatives with monoterpenoid fragments against tyrosyl-DNA phosphodiesterase 1. Russ. J. Biorgan. Chem. 2015, 41, 657–662. [Google Scholar] [CrossRef]
- Ponomarev, K.Y.; Suslov, E.V.; Zakharenko, A.L.; Zakharova, O.D.; Rogachev, A.D.; Korchagina, D.V.; Zafar, A.; Reynisson, J.; Nefedov, A.A.; Volcho, K.P. Aminoadamantanes containing monoterpene-derived fragments as potent tyrosyl-DNA phosphodiesterase 1 inhibitors. Biorgan. Chem. 2018, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.L.; Mozhaitsev, E.S.; Suslov, E.V.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F.; Lavrik, O.I. Synthesis and Inhibitory Properties of Imines Containing Monoterpenoid and Adamantane Fragments Against DNA Repair Enzyme Tyrosyl-DNA Phosphodiesterase 1 (Tdp1). Chem. Nat. Compd. 2018, 54, 672–676. [Google Scholar] [CrossRef]
- Mozhaitsev, E.S.; Zakharenko, A.L.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Vasil’eva, I.A.; Chepanova, A.A.; Black, E.; Patel, J.; Chand, R. Novel Inhibitors of DNA Repair Enzyme TDP1 Combining Monoterpenoid and Adamantane Fragments. Anticancer Agents Med. Chem. 2018, 19, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Akgun, B.; Hall, D.G. Fast and Tight Boronate Formation for Click Bioorthogonal Conjugation. Angew. Chem. Int. Ed. 2016, 55, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Reding, M.T.; Kaburagi, Y.; Tokuyama, H. Synthesis of 2,3-Disubstituted Indoles by Radical Cyclization with Hypophosphorous Acid and Its Application to Total Synthesis of (±) -Catharanthine. Heterocycles 2002, 56, 313. [Google Scholar] [CrossRef]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G.J. Insights into Substrate Binding and Catalytic Mechanism of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) from Vanadate and Tungstate-inhibited Structures. J. Mol. Biol. 2002, 324, 917–932. [Google Scholar] [CrossRef]
- Zhu, F.; Logan, G.; Reynisson, J. Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study. Mol. Inform. 2012, 31, 847–855. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Scigress, version FJ 2.6 (EU 3.1.7); Fujitsu Limited: Tokyo, Japan, 2008–2016.
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided. Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Bioinform. 2005, 61, 272–287. [Google Scholar] [CrossRef] [PubMed]
- QikProp, version 3.2; Schrödinger: New York, NY, USA, 2009.
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Interthal, H.; Pouliot, J.J.; Champoux, J.J. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. USA. 2001, 98, 12009–12014. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Huang, R.; Ayine-Tora, D.M.; Muhammad Rosdi, M.N.; Li, Y.; Reynisson, J.; Leung, I.K.H. Virtual screening and biophysical studies lead to HSP90 inhibitors. Biorgan. Med. Chem. Lett. 2017, 27, 277–281. [Google Scholar] [CrossRef]
- Molle, G.; Bauer, P.; Dubois, J.E. Formation of cage-structure organomagnesium compounds. Influence of the degree of adsorption of the transient species at the metal surface. J. Org. Chem. 1982, 47, 4120–4128. [Google Scholar] [CrossRef]
- Madder, A.; Sebastian, S.; Van Haver, D.; De Clercq, P.J.; Maskill, H. Mechanism of esterification of 1,3-dimethylamino alcohols by N-acetylimidazole in acetonitrile and the influence of alkyl and geminal dialkyl substitution upon the rate. J. Chem. Soc. Perkin Trans. 1997, 2, 2787–2793. [Google Scholar] [CrossRef]
- Mukherjee, A.; Wu, Q.; le Noble, W.J. Face Selection in Claisen Rearrangements. J. Org. Chem. 1994, 59, 3270–3274. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.W.; Falconi, M.; Kristoffersen, E.L.; Simonsen, A.T.; Cifuentes, J.B.; Marcussen, L.B.; Frøhlich, R.; Vagner, J.; Harmsen, C.; Juul, S. Real-time detection of TDP1 activity using a fluorophore-quencher coupled DNA-biosensor. Biosens. Bioelectron. 2013, 48, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Martino, E.; Della Volpe, S.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Biorgan. Med. Chem. Lett. 2017, 27, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inform. 2019, 38, 1800068–1800078. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Structure | IC50, μM | ||
|---|---|---|---|---|
| R1 | R2 | X | ||
| 44a |  |  | O | >75 |
| 46a |  |  | S | 3.3 ± 0.5 (2.8–3.8) * |
| 45a |  |  | O | 5.2 ± 0.2 (5.0–5.4) * |
| 47a |  |  | S | 0.64 ± 0.17 (0.47–0.81) * |
| 44b |  |  | O | >75 |
| 45b |  |  | O | 2.5 ± 0.1 (2.4–2.6) * |
| 50a |  |  | O | >75 |
| 50b |  |  | O | 13.4 ± 3.0 (10.4–16.4) * |
| 51 |  |  | S | 2.3 ± 0.8 (1.5–3.1) * |
| 11 | Furamidine | 1.23 ± 0.33 (0.90–1.56) * | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chepanova, A.A.; Mozhaitsev, E.S.; Munkuev, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Zakharenko, A.L.; Patel, J.; Ayine-Tora, D.M.; Reynisson, J.; et al. The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Appl. Sci. 2019, 9, 2767. https://doi.org/10.3390/app9132767
Chepanova AA, Mozhaitsev ES, Munkuev AA, Suslov EV, Korchagina DV, Zakharova OD, Zakharenko AL, Patel J, Ayine-Tora DM, Reynisson J, et al. The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Applied Sciences. 2019; 9(13):2767. https://doi.org/10.3390/app9132767
Chicago/Turabian StyleChepanova, Arina A., Evgenii S. Mozhaitsev, Aldar A. Munkuev, Evgeniy V. Suslov, Dina V. Korchagina, Olga D. Zakharova, Alexandra L. Zakharenko, Jinal Patel, Daniel M. Ayine-Tora, Jóhannes Reynisson, and et al. 2019. "The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges" Applied Sciences 9, no. 13: 2767. https://doi.org/10.3390/app9132767
APA StyleChepanova, A. A., Mozhaitsev, E. S., Munkuev, A. A., Suslov, E. V., Korchagina, D. V., Zakharova, O. D., Zakharenko, A. L., Patel, J., Ayine-Tora, D. M., Reynisson, J., Leung, I. K. H., Volcho, K. P., Salakhutdinov, N. F., & Lavrik, O. I. (2019). The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Applied Sciences, 9(13), 2767. https://doi.org/10.3390/app9132767