Quinoxaline-Based Dual Donor, Dual Acceptor Organic Dyes for Dye-Sensitized Solar Cells

 , ,
, ,  and
and
Abstract
Featured Application
Abstract
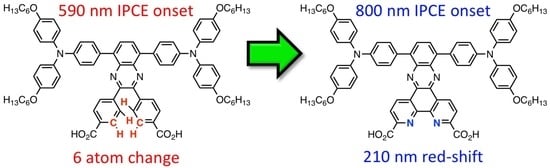
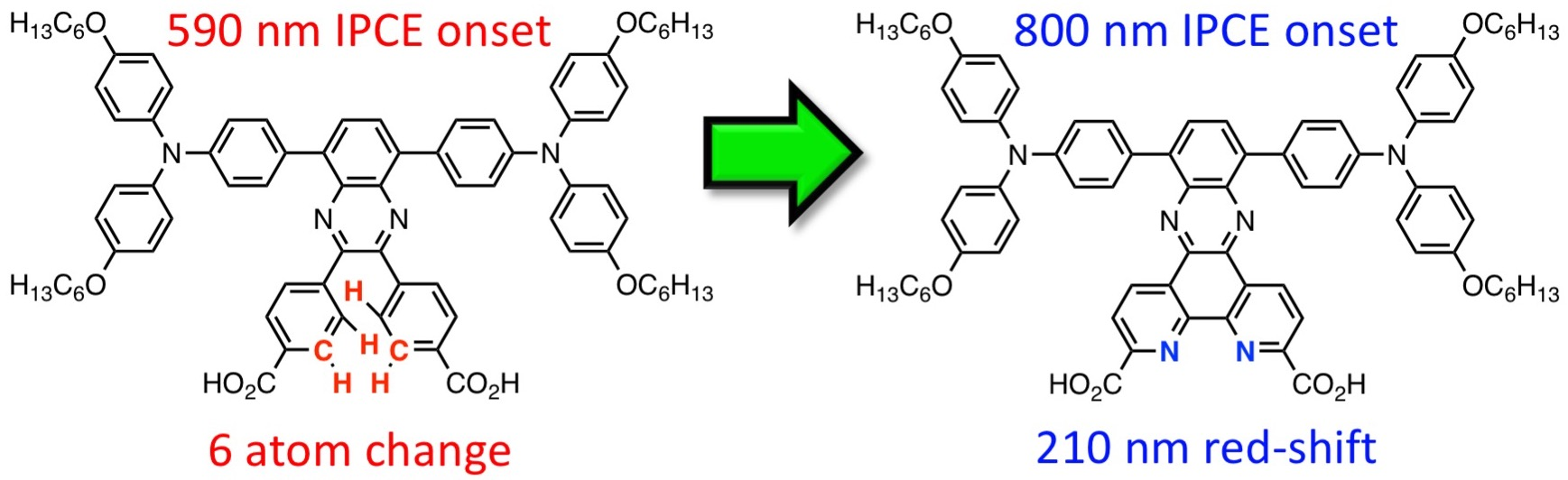
1. Introduction
2. Materials and Methods
Known Synthetic Intermediates
3. Result and Discussion
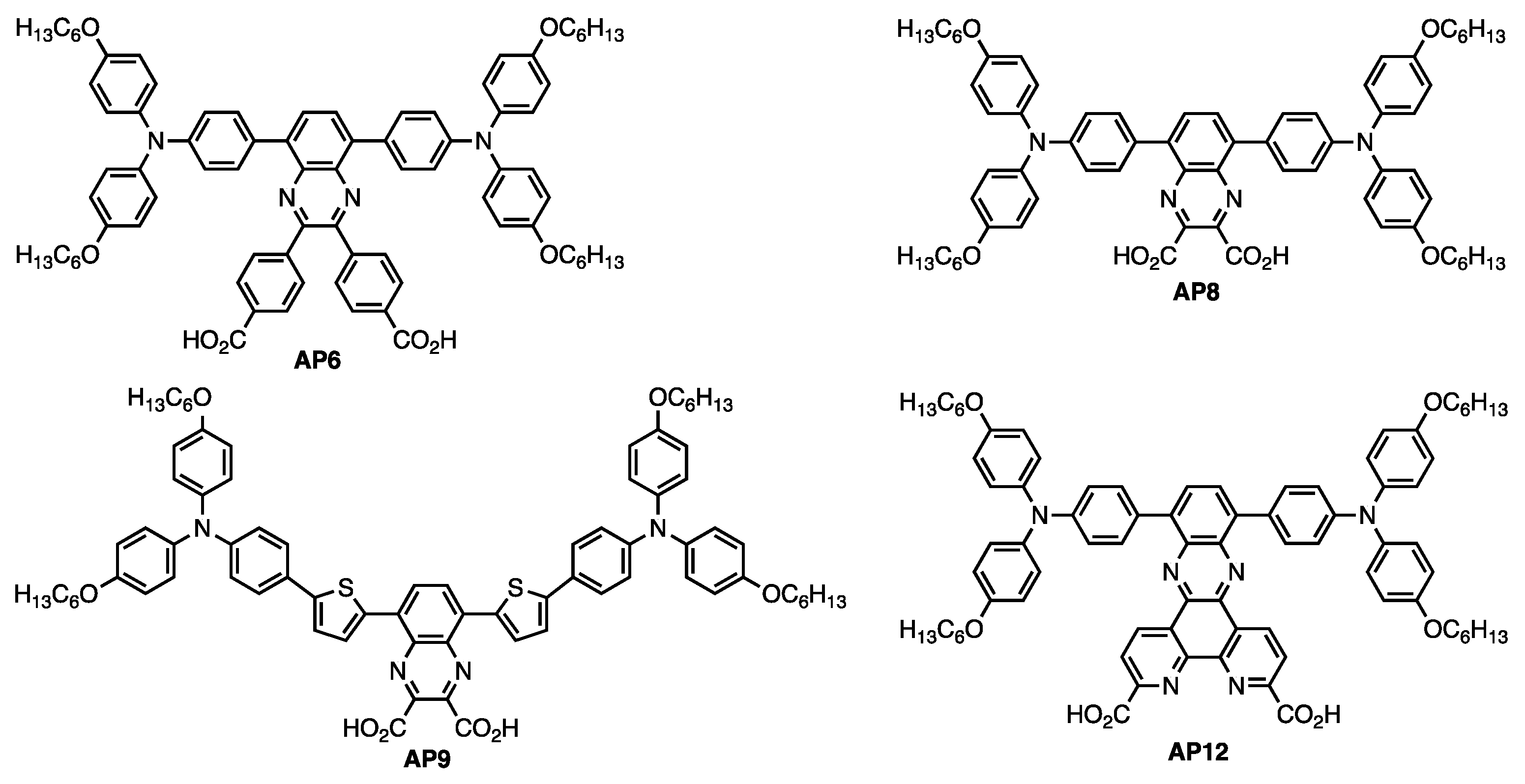
3.1. AP9 and AP12 Synthesis Discussion
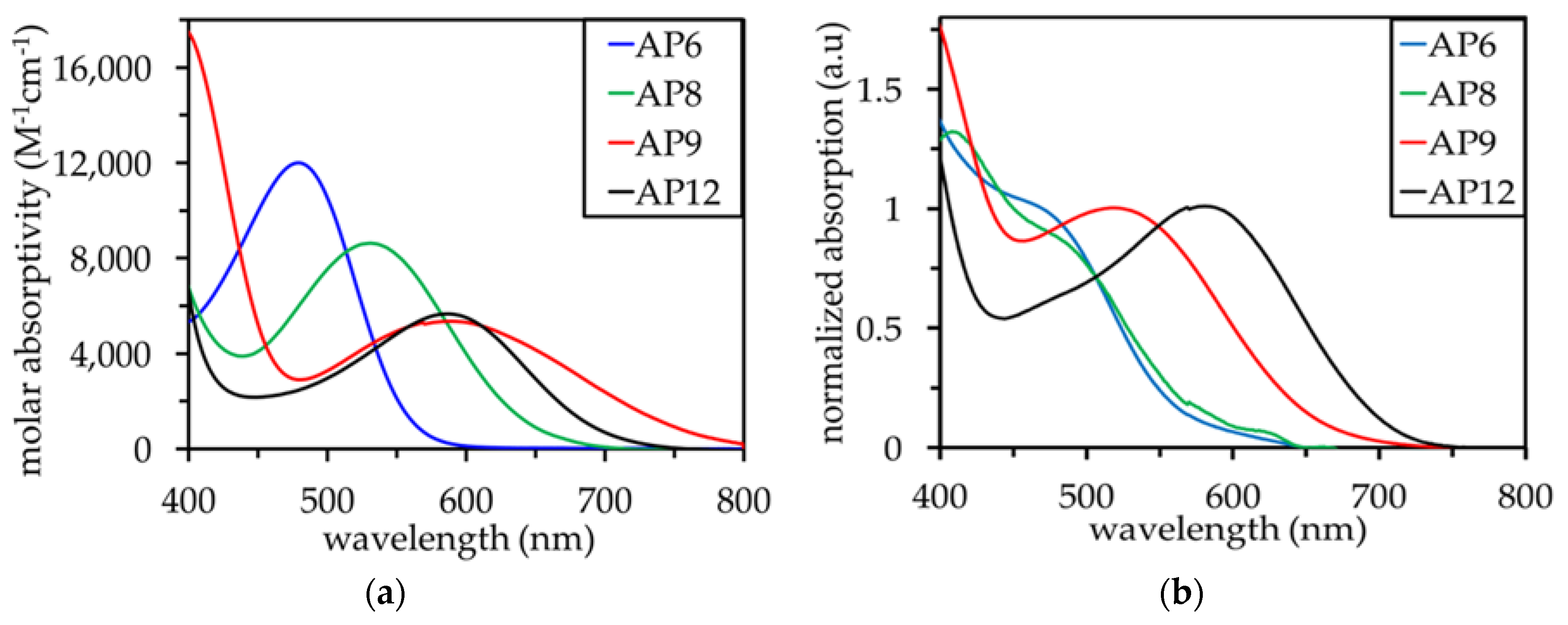
3.2. UV–Vis Absorption Properties
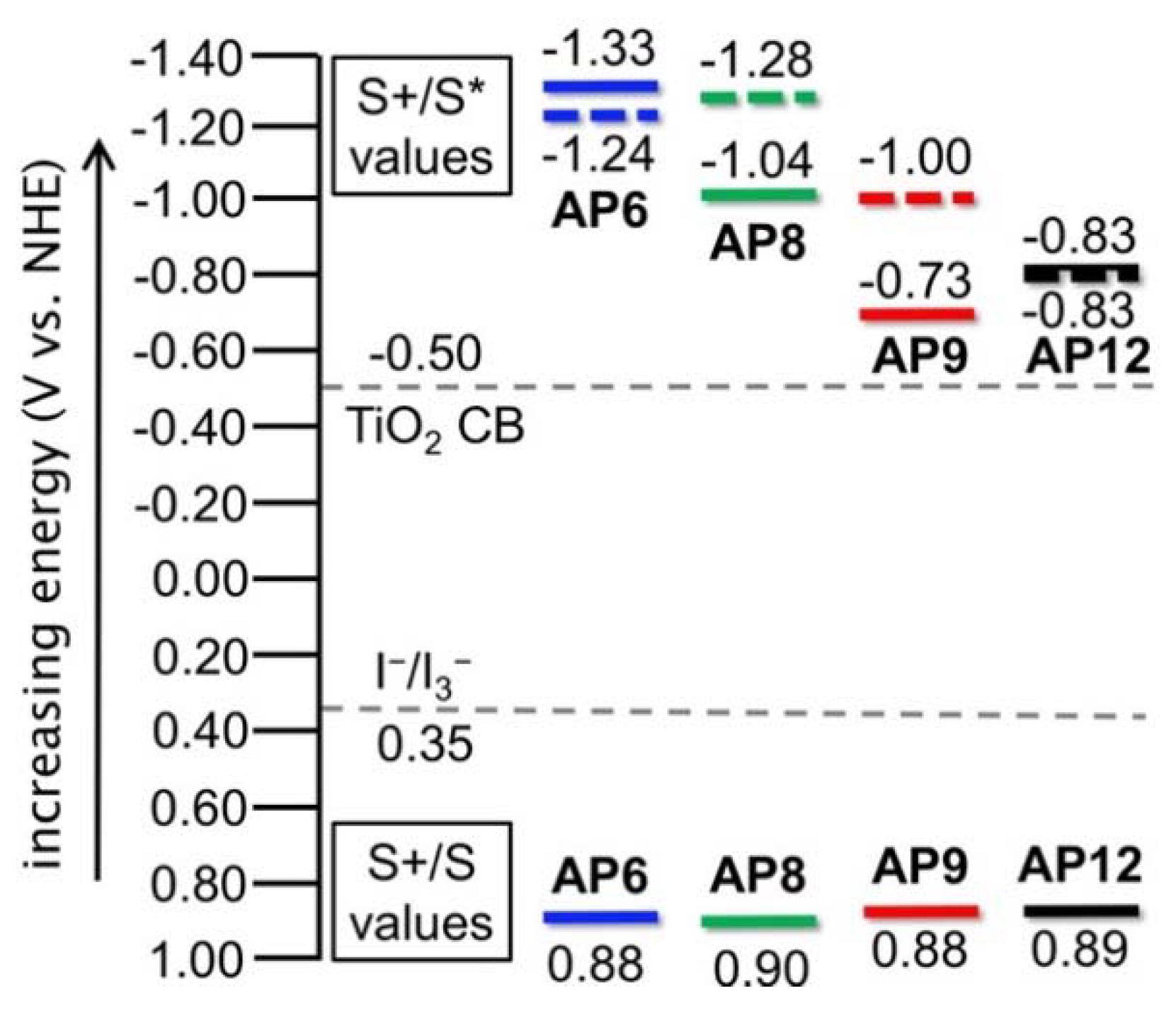
3.3. Electrochemical Properties
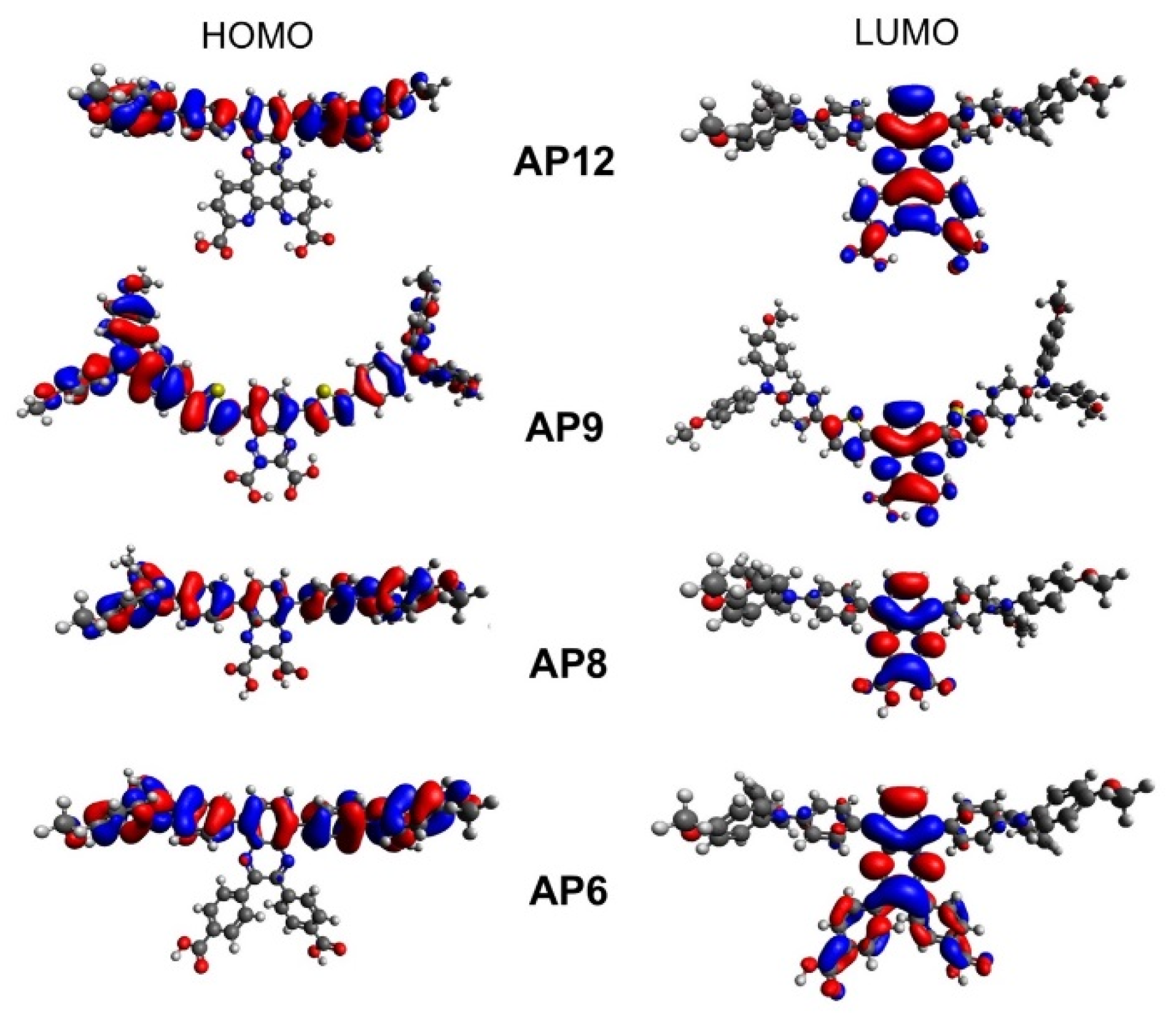
3.4. Computational Analysis
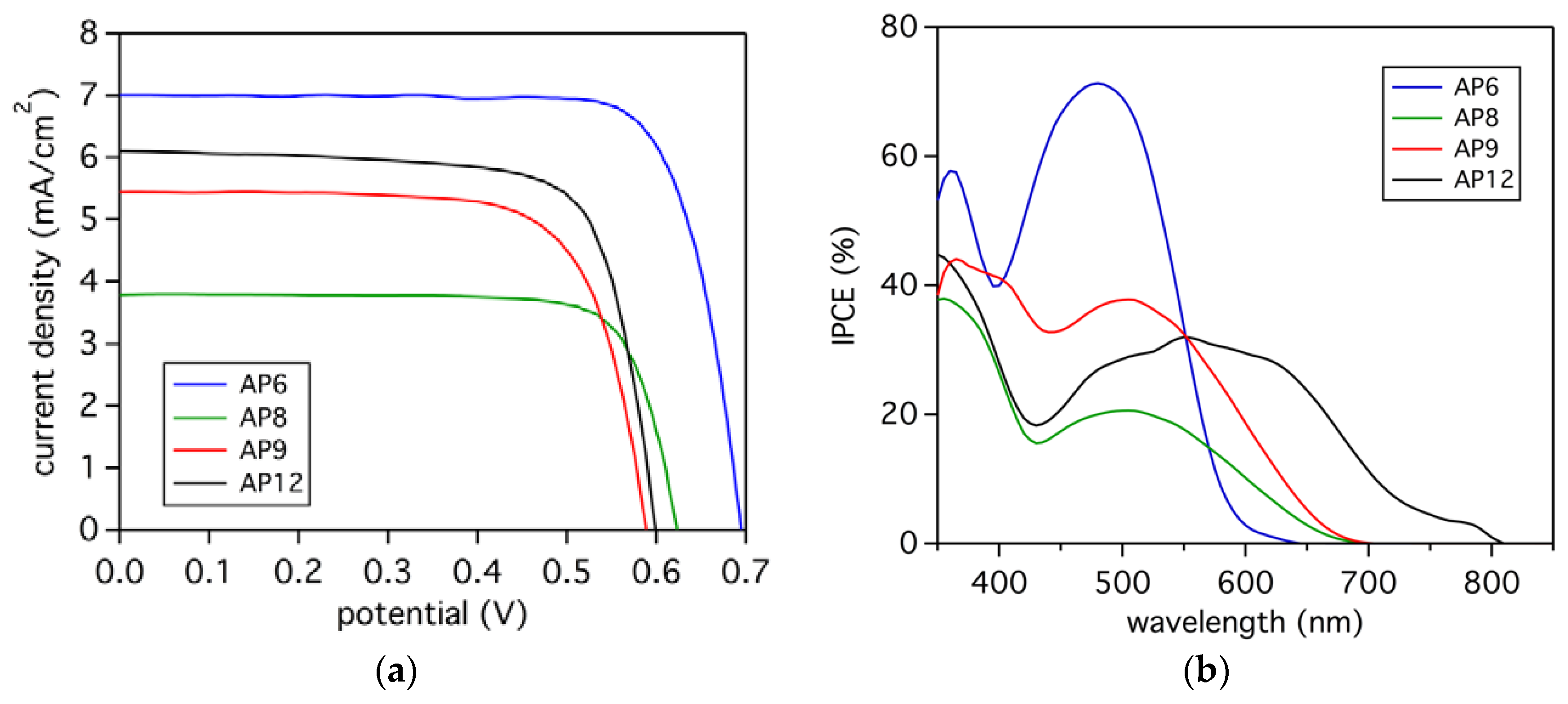
3.5. Photovoltaic Analysis
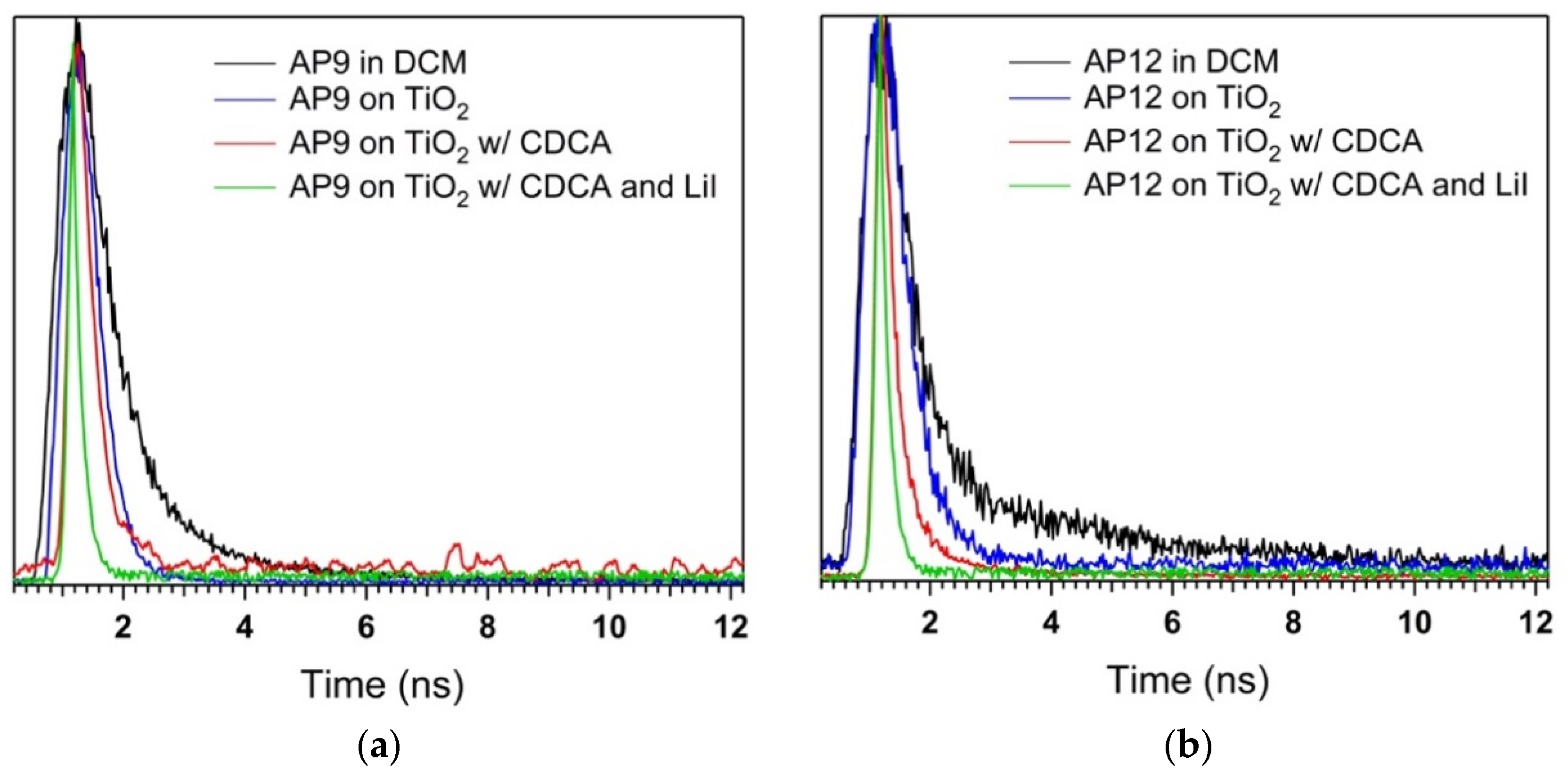
3.6. Time-Correlated Single Photon Counting (TCSPC) Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boschloo, G.; Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, B.C.; Durrant, J.R. Kinetic and energetic paradigms for dye-sensitized solar cells: Moving from the ideal to the real. Acc. Chem. Res. 2009, 42, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liang, M.; Chen, J. Kinetics of iodine-free redox shuttles in dye-sensitized solar cells: Interfacial recombination and dye regeneration. Acc. Chem. Res. 2015, 48, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Fischer, M.K.; Bauerle, P. Metal-free organic dyes for dye-sensitized solar cells: From structure: Property relationships to design rules. Angew. Chem. Int. Ed. 2009, 48, 2474–2499. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in dye-sensitized solar cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Ardo, S.; Meyer, G.J. Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces. Chem. Soc. Rev. 2009, 38, 115–164. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Gerbaldi, C.; Barolo, C.; Gratzel, M. Aqueous dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3431–3473. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Guillén, E.; Kavan, L.; Grätzel, M.; Nazeeruddin, M.K. Metal free sensitizer and catalyst for dye sensitized solar cells. Energy Environ. Sci. 2013, 6, 3439–3466. [Google Scholar] [CrossRef]
- Fakharuddin, A.; Jose, R.; Brown, T.M.; Fabregat-Santiago, F.; Bisquert, J. A perspective on the production of dye-sensitized solar modules. Energy Environ. Sci. 2014, 7, 3952–3981. [Google Scholar] [CrossRef]
- Hamann, T.W.; Ondersma, J.W. Dye-sensitized solar cell redox shuttles. Energy Environ. Sci. 2011, 4, 370–381. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Numata, Y.; Han, L. Highly efficient dye-sensitized solar cells: Progress and future challenges. Energy Environ. Sci. 2013, 6, 1443–1464. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Wenger, S.; Nazeeruddin, M.K.; Grätzel, M. Panchromatic engineering for dye-sensitized solar cells. Energy Environ. Sci. 2011, 4, 842–857. [Google Scholar] [CrossRef]
- Pashaei, B.; Shahroosvand, H.; Abbasi, P. Transition metal complex redox shuttles for dye-sensitized solar cells. RSC Adv. 2015, 5, 94814–94848. [Google Scholar] [CrossRef]
- Polman, A.; Knight, M.; Garnett, E.C.; Ehrler, B.; Sinke, W.C. Photovoltaic materials: Present efficiencies and future challenges. Science 2016, 352, aad4424. [Google Scholar] [CrossRef] [PubMed]
- Hardin, B.E.; Snaith, H.J.; McGehee, M.D. The renaissance of dye-sensitized solar cells. Nat. Photonics 2012, 6, 162–169. [Google Scholar] [CrossRef]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.I.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, W.H.; Zakeeruddin, S.M.; Gratzel, M. Insight into D-A-π-A structured sensitizers: A promising route to highly efficient and stable dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2015, 7, 9307–9318. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Chen, J. Arylamine organic dyes for dye-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 3453–3488. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Eom, Y.K.; Kang, S.H.; Choi, I.T.; Yoo, Y.; Kim, J.; Kim, H.K. Significant light absorption enhancement by a single heterocyclic unit change in the π-bridge moiety from thieno[3,2-b]benzothiophene to thieno[3,2-b]indole for high performance dye-sensitized and tandem solar cells. J. Mater. Chem. A 2017, 5, 2297–2308. [Google Scholar] [CrossRef]
- Sobus, J.; Gierczyk, B.; Burdzinski, G.; Jancelewicz, M.; Polanski, E.; Hagfeldt, A.; Ziolek, M. Factors affecting the performance of champion silyl-anchor carbazole dye revealed in the femtosecond to second studies of complete ADEKA-1 sensitized solar cells. Chem. Eur. J. 2016, 22, 15807–15818. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, N.; Cecconi, B.; Abbotto, A. Multi-branched multi-anchoring metal-free dyes for dye-sensitized solar cells. Eur. J. Org. Chem. 2014, 2014, 7069–7086. [Google Scholar] [CrossRef]
- Brogdon, P.; McNamara, L.E.; Peddapuram, A.; Hammer, N.I.; Delcamp, J.H. Toward tightly bound carboxylic acid-based organic dyes for dscs: Relative TiO2 binding strengths of benzoic acid, cyanoacrylic acid, and conjugated double carboxylic acid anchoring dyes. Synth. Met. 2016, 222, 66–75. [Google Scholar] [CrossRef]
- Peddapuram, A.; Cheema, H.; Adams, R.E.; Schmehl, R.H.; Delcamp, J.H. A stable panchromatic green dual acceptor, dual donor organic dye for dye-sensitized solar cells. J. Phys. Chem. C 2017, 121, 8770–8780. [Google Scholar] [CrossRef]
- Cheema, H.; Peddapuram, A.; Adams, R.E.; McNamara, L.E.; Hunt, L.A.; Le, N.; Watkins, D.L.; Hammer, N.I.; Schmehl, R.H.; Delcamp, J.H. Molecular engineering of NIR absorbing thienopyrazine double donor double acceptor organic dyes for DSCs. J. Org. Chem. 2017, 82, 12038–12049. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, N.P.; Yella, A.; Nazeeruddin, M.; Grätzel, M.; Delcamp, J.H. Thieno[3,4-b]pyrazine as an electron deficient π-bridge in D-A-π-A DSCs. ACS Appl. Mater. Interfaces 2016, 8, 5376–5384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Li, G.; Zhan, X.; Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics 2018, 12, 131–142. [Google Scholar] [CrossRef]
- Cheng, P.; Zhan, X. Stability of organic solar cells: Challenges and strategies. Chem. Soc. Rev. 2016, 45, 2544–2582. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Watanabe, M.; Chou, P.T.; Chow, T.J. [2.2]paracyclophane as a bridging unit in the design of organic dyes for sensitized solar cells. Chem. Commun. 2012, 48, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tsang, S.W.; Zhang, W.; Tao, Y.; Wong, M.S. Naphthodithiophene-2,1,3-benzothiadiazole copolymers for bulk heterojunction solar cells. Chem. Commun. 2011, 47, 9471–9473. [Google Scholar] [CrossRef] [PubMed]
- Mátravölgyi, B.; Hergert, T.; Thurner, A.; Varga, B.; Sangiorgi, N.; Bendoni, R.; Zani, L.; Reginato, G.; Calamante, M.; Sinicropi, A.; et al. Synthesis and investigation of solar-cell photosensitizers having a fluorazone backbone. Eur. J. Org. Chem. 2017, 2017, 1843–1854. [Google Scholar] [CrossRef]
- Gislason, K.; Sigurdsson, S.T. Rigid 5′-6-locked phenanthroline-derived nucleosides chelated to ruthenium and europium ions. Bioorg. Med. Chem. Lett. 2013, 23, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scurseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, A.; et al. Gaussian09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Brogdon, P.; Cheema, H.; Delcamp, J.H. Near-infrared-absorbing metal-free organic, porphyrin, and phthalocyanine sensitizers for panchromatic dye-sensitized solar cells. ChemSusChem 2018, 11, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-R.; Lee, C.-P.; Liao, C.-W.; Su, W.-L.; Li, C.-T.; Ho, K.-C.; Sun, S.-S. Structural engineering of dipolar organic dyes with an electron-deficient diphenylquinoxaline moiety for efficient dye-sensitized solar cells. Tetrahedron 2014, 70, 6276–6284. [Google Scholar] [CrossRef]
- Falgenhauer, J.; Richter, C.; Miura, H.; Schlettwein, D. Stable sensitization of ZnO by improved anchoring of indoline dyes. ChemPhysChem 2012, 13, 2893–2897. [Google Scholar] [CrossRef] [PubMed]
- Aghazada, S.; Nazeeruddin, M. Ruthenium complexes as sensitizers in dye-sensitized solar cells. Inorganics 2018, 6, 52. [Google Scholar] [CrossRef]
- Baumann, A.; Cheema, H.; Sabuj, M.A.; McNamara, L.E.; Zhang, Y.; Peddapuram, A.; Nguyen, S.T.; Watkins, D.L.; Hammer, N.I.; Rai, N.; et al. Iodine binding with thiophene and furan based dyes for DSCs. Phys. Chem. Chem. Phys. 2018, 20, 17859–17870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheema, H.; McNamara, L.E.; Hunt, L.A.; Hammer, N.I.; Delcamp, J.H. Ullazine donor-π bridge-acceptor organic dyes for dye-sensitized solar cells. Chem. Eur. J. 2018, 24, 5939–5949. [Google Scholar] [CrossRef] [PubMed]
- Huckaba, A.J.; Giordano, F.; McNamara, L.E.; Dreux, K.M.; Hammer, N.I.; Tschumper, G.S.; Zakeeruddin, S.M.; Grätzel, M.; Nazeeruddin, M.K.; Delcamp, J.H. Indolizine-based donors as organic sensitizer components for dye-sensitized solar cells. Adv. Energy Mater. 2015, 5, 1401629. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorbance Data [a] | Electrochemical Data | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dye | λmax (nm) | λonsetDCM (nm) | λonsetTiO2 (nm) | IPCEonset (nm) | ε (M−1 cm−1) | E(S+/S) DCM | Egopt (eV) [b] | E(S+/S*) DCM [c] | E(S+/S*) TiO2 [c] | E(S+/S*) IPCE [c] |
| AP6 | 479 | 560 | 585 | 590 | 12,000 | 0.88 | 2.21 | −1.33 | −1.24 | −1.22 |
| AP8 | 531 | 640 | 570 | 675 | 8,000 | 0.90 | 1.94 | −1.04 | −1.28 | −0.94 |
| AP9 | 590 | 770 | 660 | 680 | 5,500 | 0.88 | 1.61 | −0.73 | −1.00 | −0.94 |
| AP12 | 588 | 720 | 720 | 800 | 5,600 | 0.89 | 1.72 | −0.83 | −0.83 | −0.66 |
| Dye | JSC (mA/cm2) | VOC (mV) | FF | PCE (%) |
|---|---|---|---|---|
| AP6 | 7.0 | 694 | 0.76 | 3.7 |
| AP8 | 3.8 | 615 | 0.71 | 1.7 |
| AP9 | 5.4 | 591 | 0.72 | 2.3 |
| AP12 | 6.1 | 599 | 0.74 | 2.8 |
| Dye | τsol (ns) a | τTiO2 (ns) b | ηeff (%) b | τTiO2 (ns)c | ηeff (%) c | τTiO2 (ns) d | ηeff (%) d |
|---|---|---|---|---|---|---|---|
| AP6 | 0.38 | 0.30 | 29 | 0.29 | 24 | <0.15 | >61 |
| AP8 | 0.34 | 0.29 | 27 | 0.27 | 21 | <0.15 | >54 |
| AP9 | 0.77 | 0.44 | 42 | 0.32 | 58 | <0.15 | >80 |
| AP12 | 0.79 | 0.55 | 30 | 0.25 | 68 | <0.15 | >81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peddapuram, A.; Cheema, H.; McNamara, L.E.; Zhang, Y.; Hammer, N.I.; Delcamp, J.H. Quinoxaline-Based Dual Donor, Dual Acceptor Organic Dyes for Dye-Sensitized Solar Cells. Appl. Sci. 2018, 8, 1421. https://doi.org/10.3390/app8091421
Peddapuram A, Cheema H, McNamara LE, Zhang Y, Hammer NI, Delcamp JH. Quinoxaline-Based Dual Donor, Dual Acceptor Organic Dyes for Dye-Sensitized Solar Cells. Applied Sciences. 2018; 8(9):1421. https://doi.org/10.3390/app8091421
Chicago/Turabian StylePeddapuram, Adithya, Hammad Cheema, Louis E. McNamara, Yanbing Zhang, Nathan I. Hammer, and Jared H. Delcamp. 2018. "Quinoxaline-Based Dual Donor, Dual Acceptor Organic Dyes for Dye-Sensitized Solar Cells" Applied Sciences 8, no. 9: 1421. https://doi.org/10.3390/app8091421
APA StylePeddapuram, A., Cheema, H., McNamara, L. E., Zhang, Y., Hammer, N. I., & Delcamp, J. H. (2018). Quinoxaline-Based Dual Donor, Dual Acceptor Organic Dyes for Dye-Sensitized Solar Cells. Applied Sciences, 8(9), 1421. https://doi.org/10.3390/app8091421