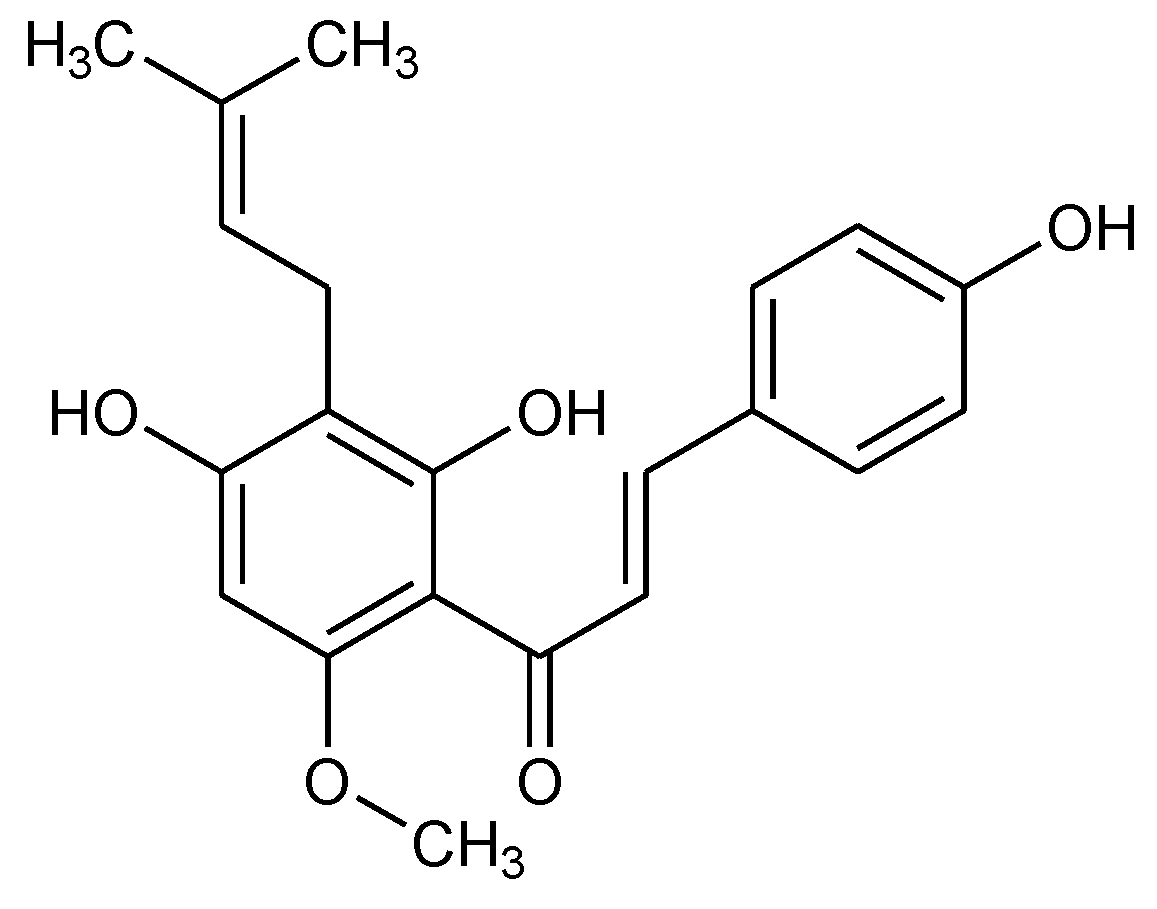
Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Cells
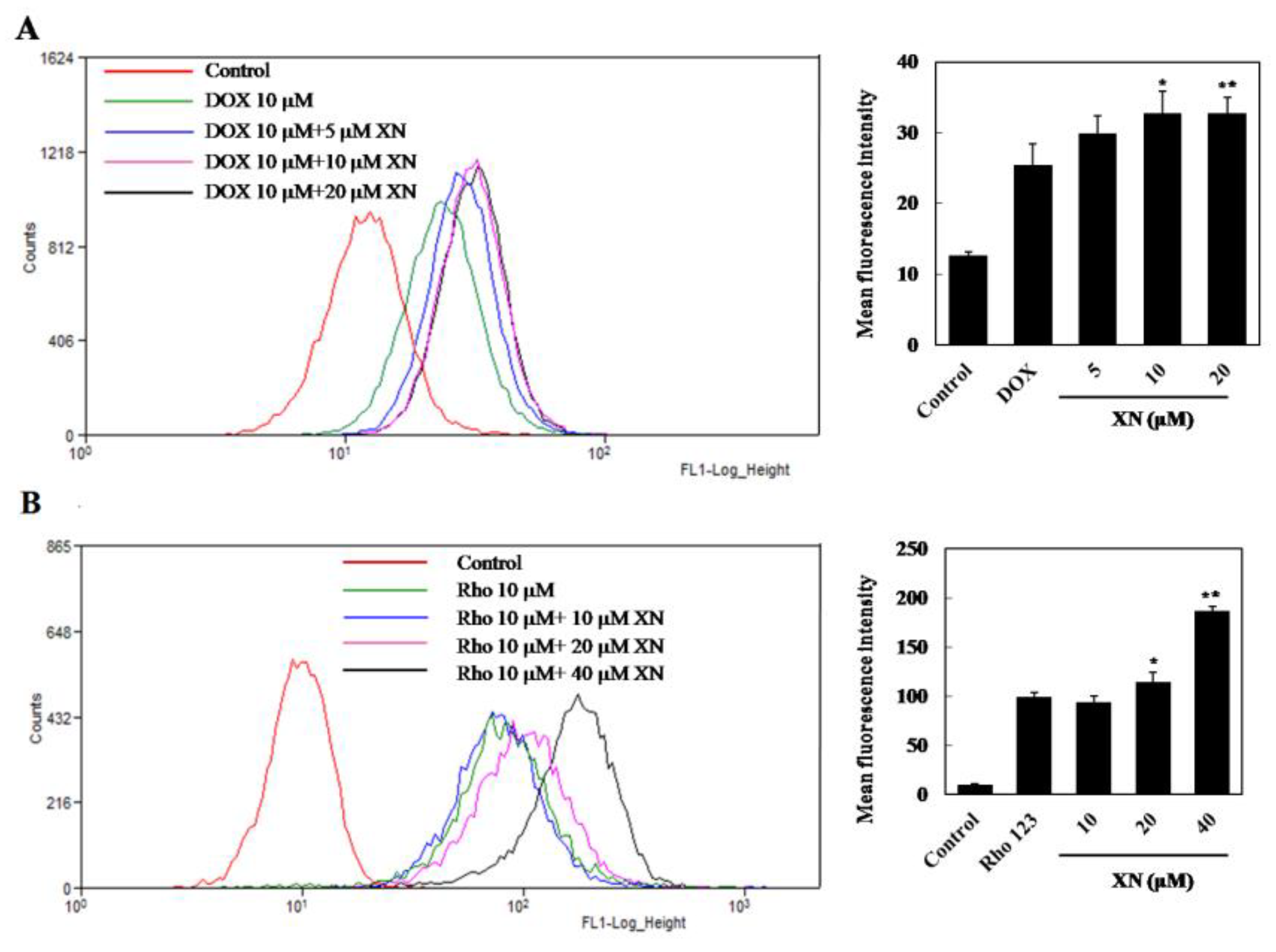
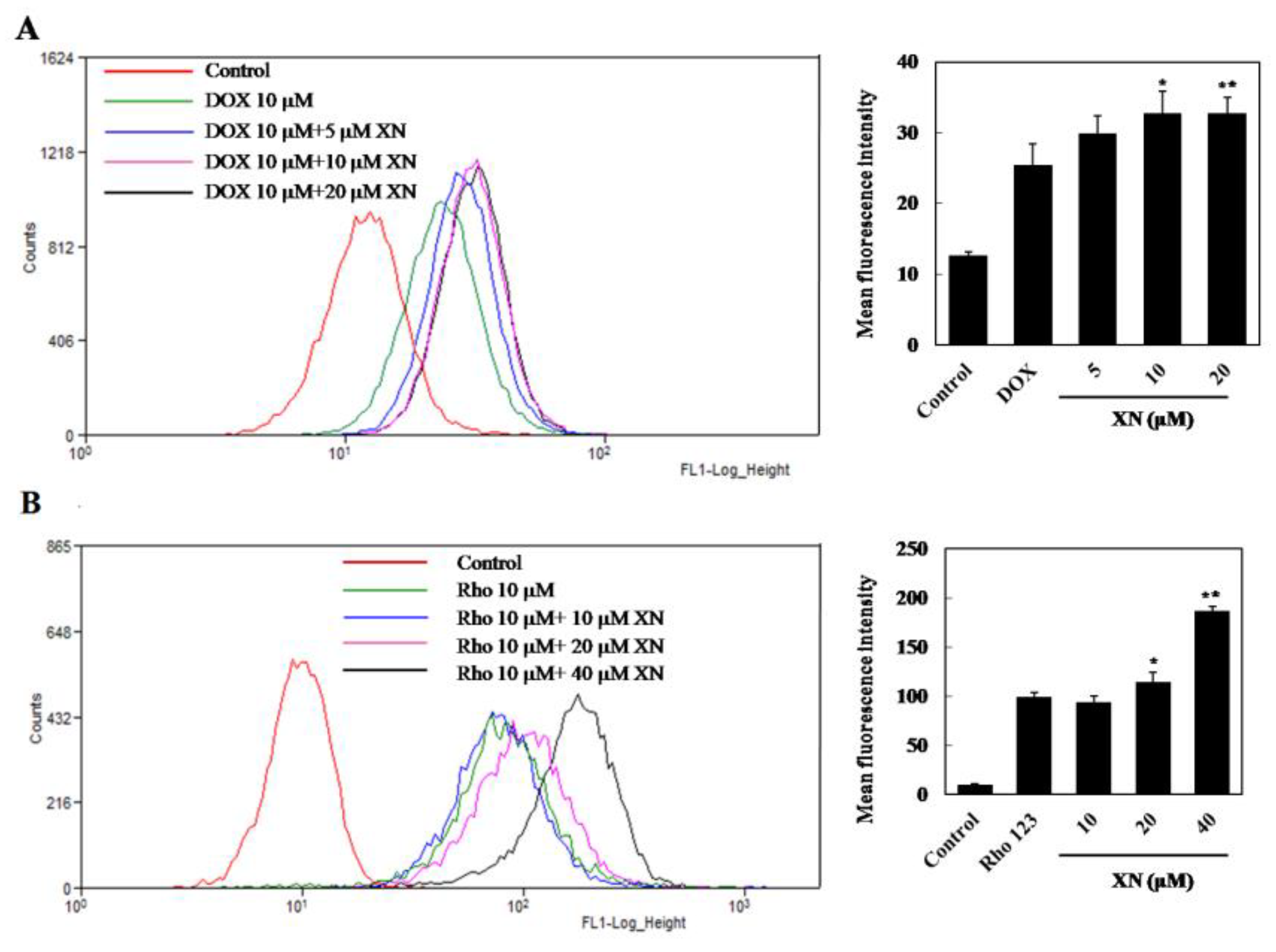
2.2. Detection of Intracellular DOX Accumulation
2.3. Rhodamine123 (Rho123) Efflux Assay
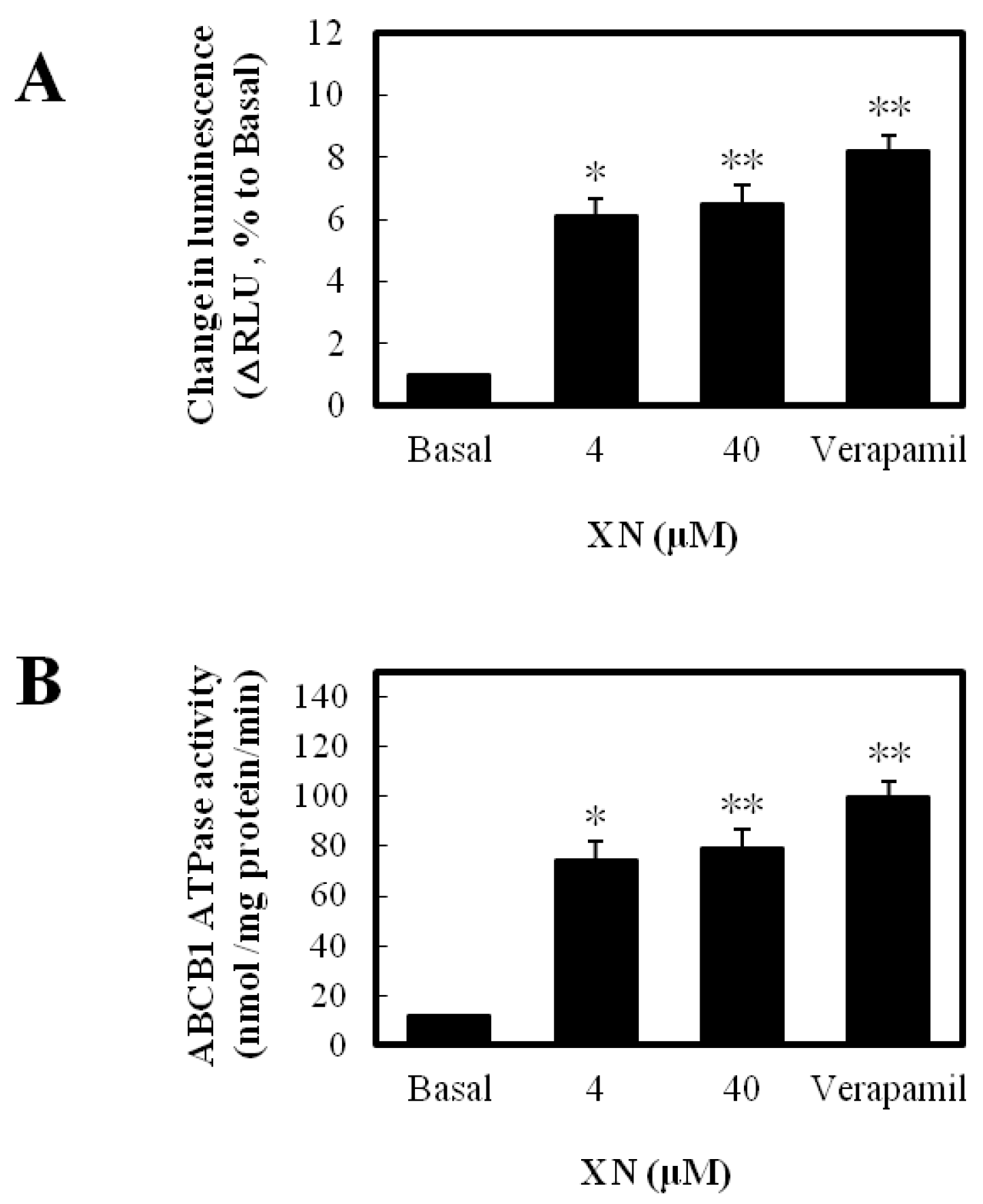
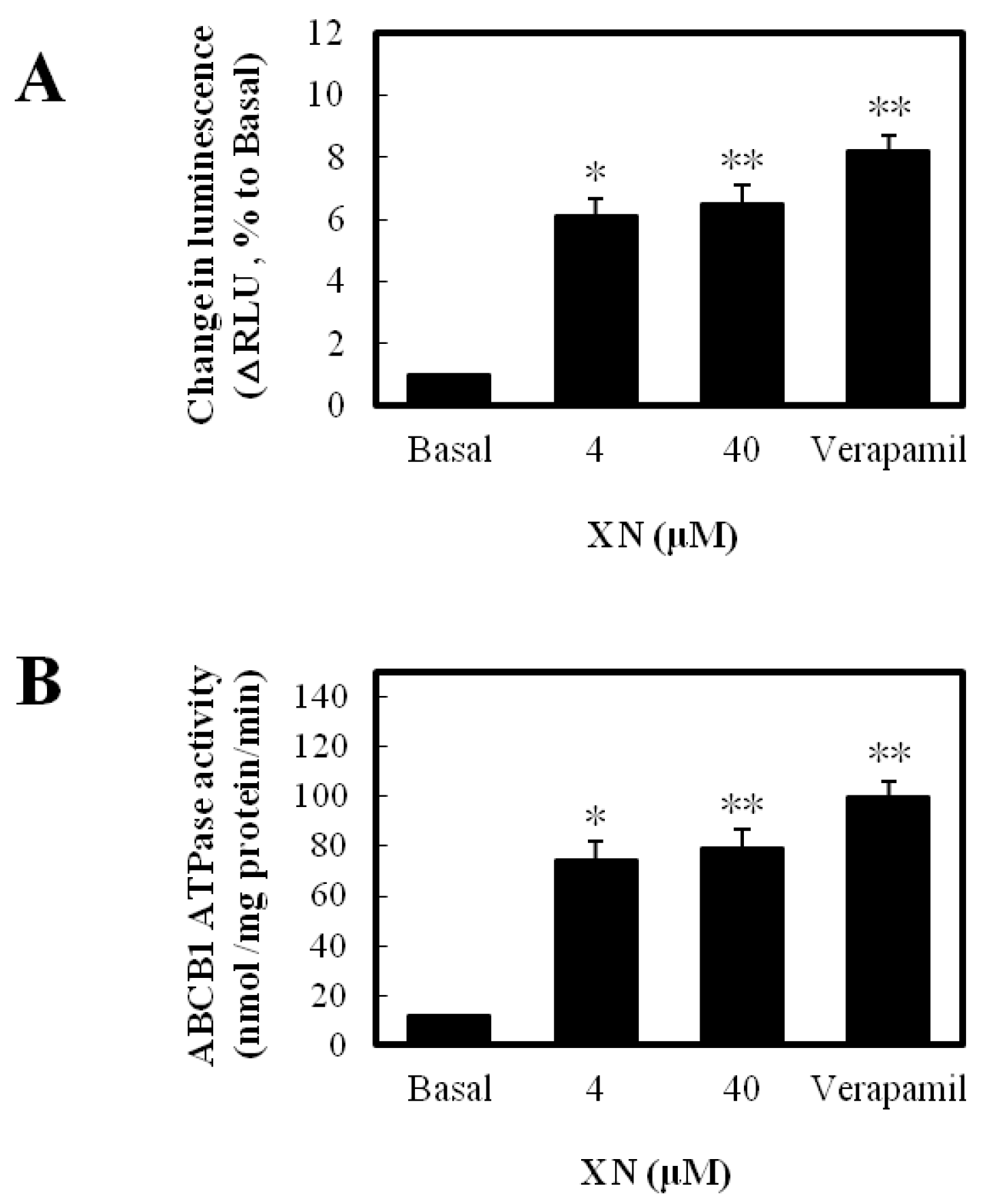
2.4. ABCB1 ATPase Activity Assay
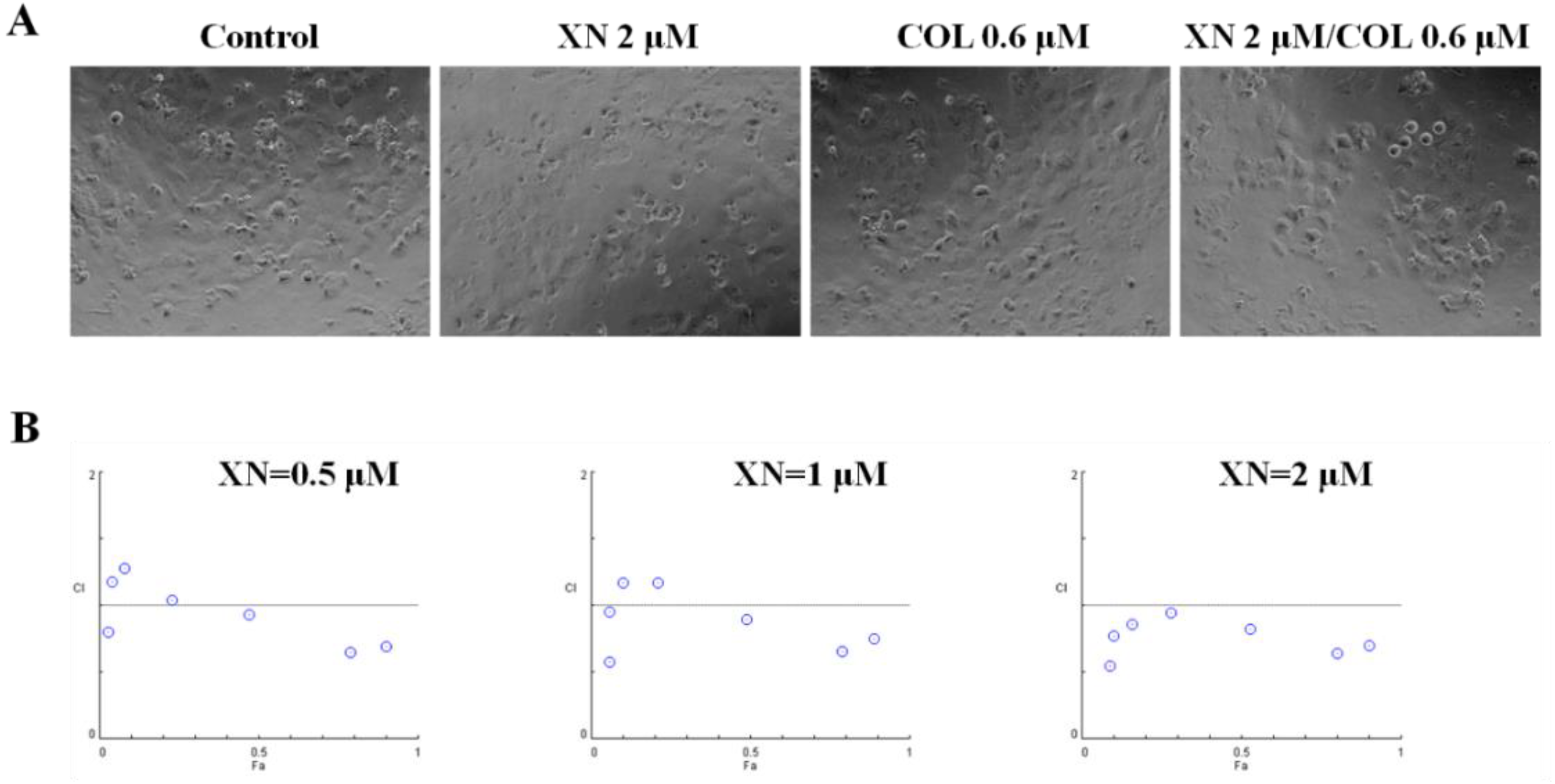
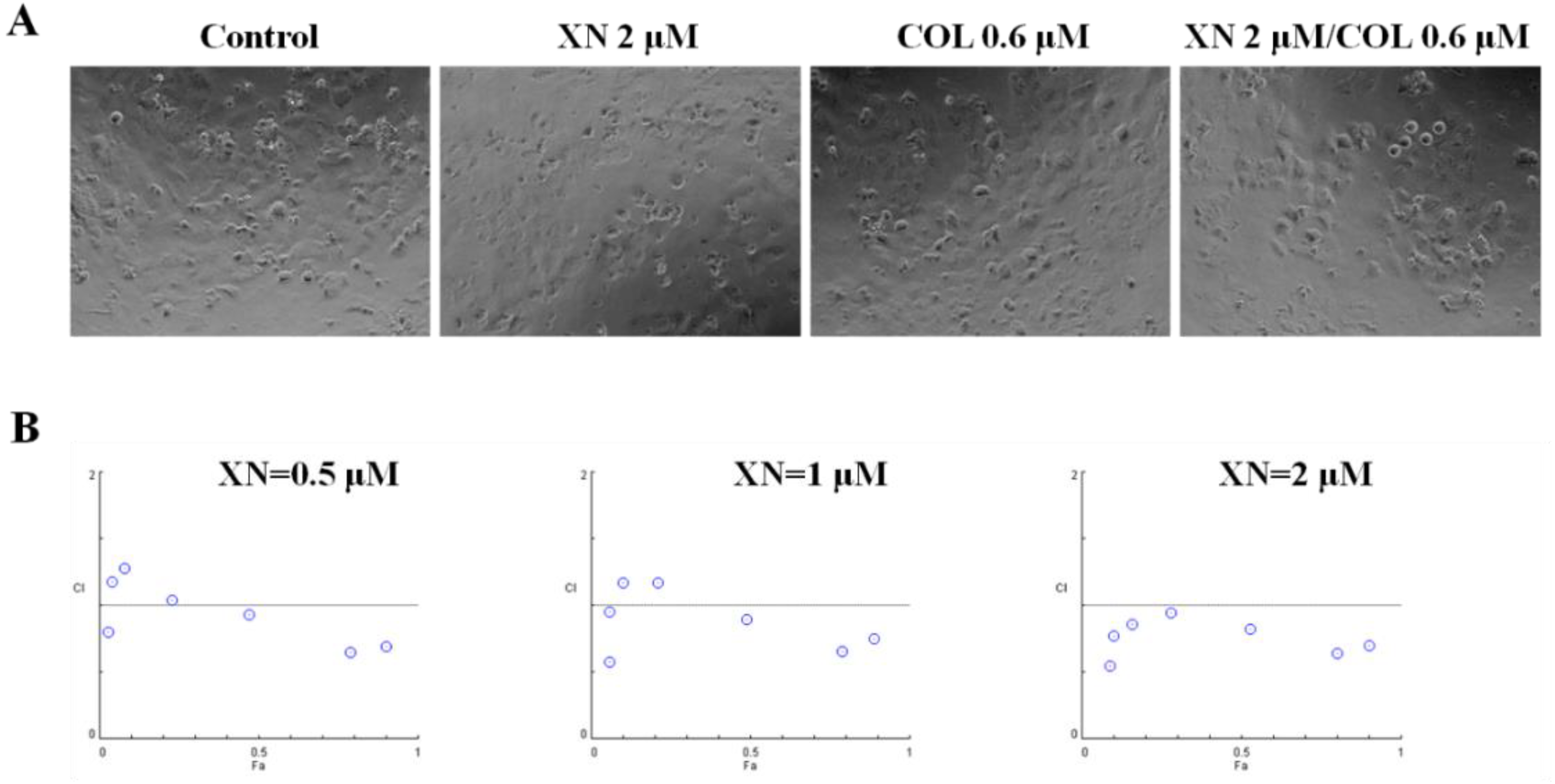
2.5. Drug Synergism Calculation
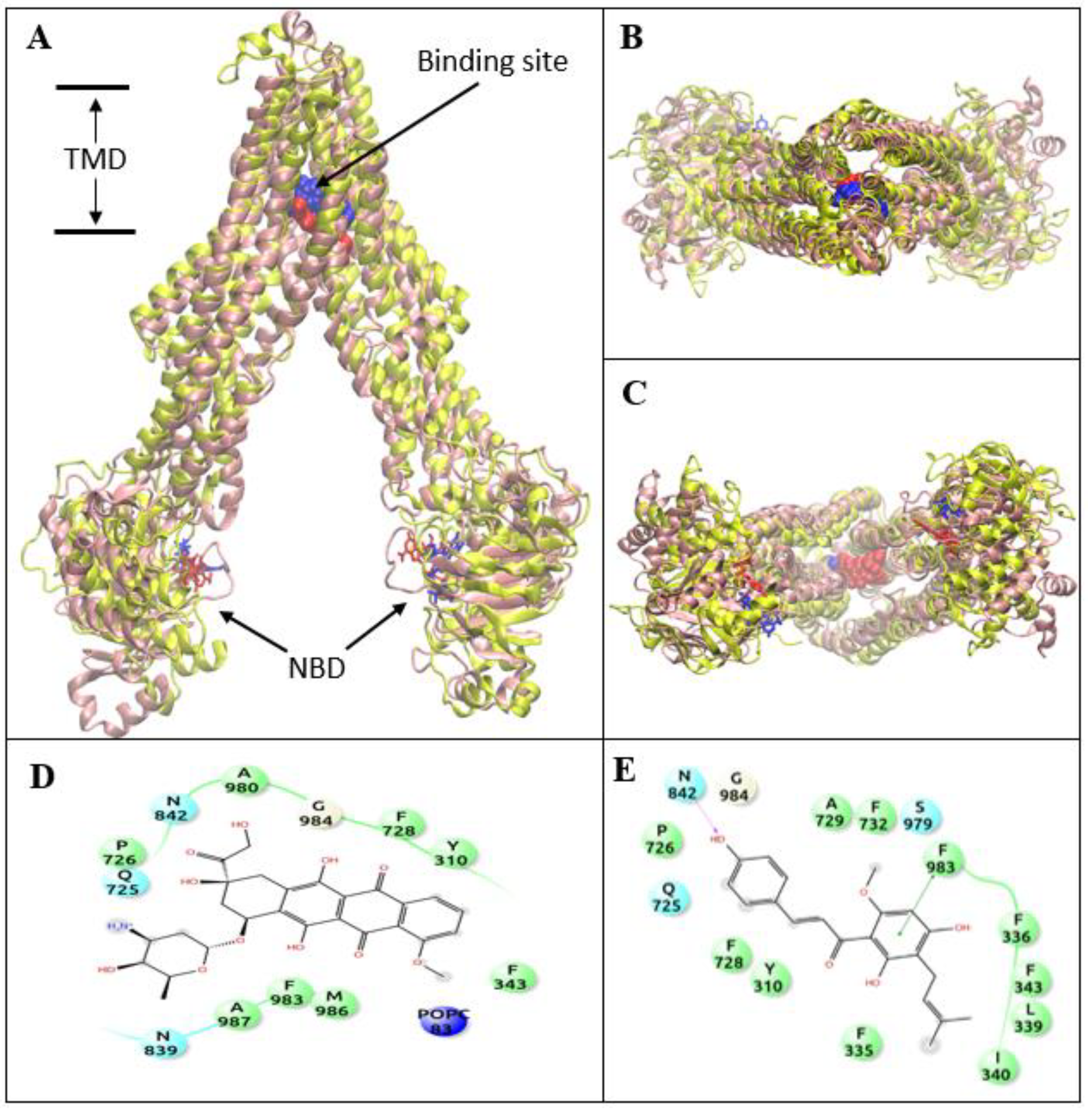
2.6. Molecular Docking
2.7. Molecular Dynamics Simulations and Analysis
2.7.1. Simulation System Setup
2.7.2. System Relaxation and MD Simulations
2.7.3. Simulation Interaction Diagrams (SID)
2.7.4. Convergence of Simulations
2.7.5. MM-GBSA Binding Energies
2.8. Data Analysis
3. Results
3.1. XN Inhibited the Efflux Functions of ABCB1 in MCF-7/ADR Cells
3.2. XN Was a Substrate of ABCB1 and Stimulated Its ATPase Activity
3.3. XN Showed a Synergic Effect with COL in MCF-7/ADR Cells
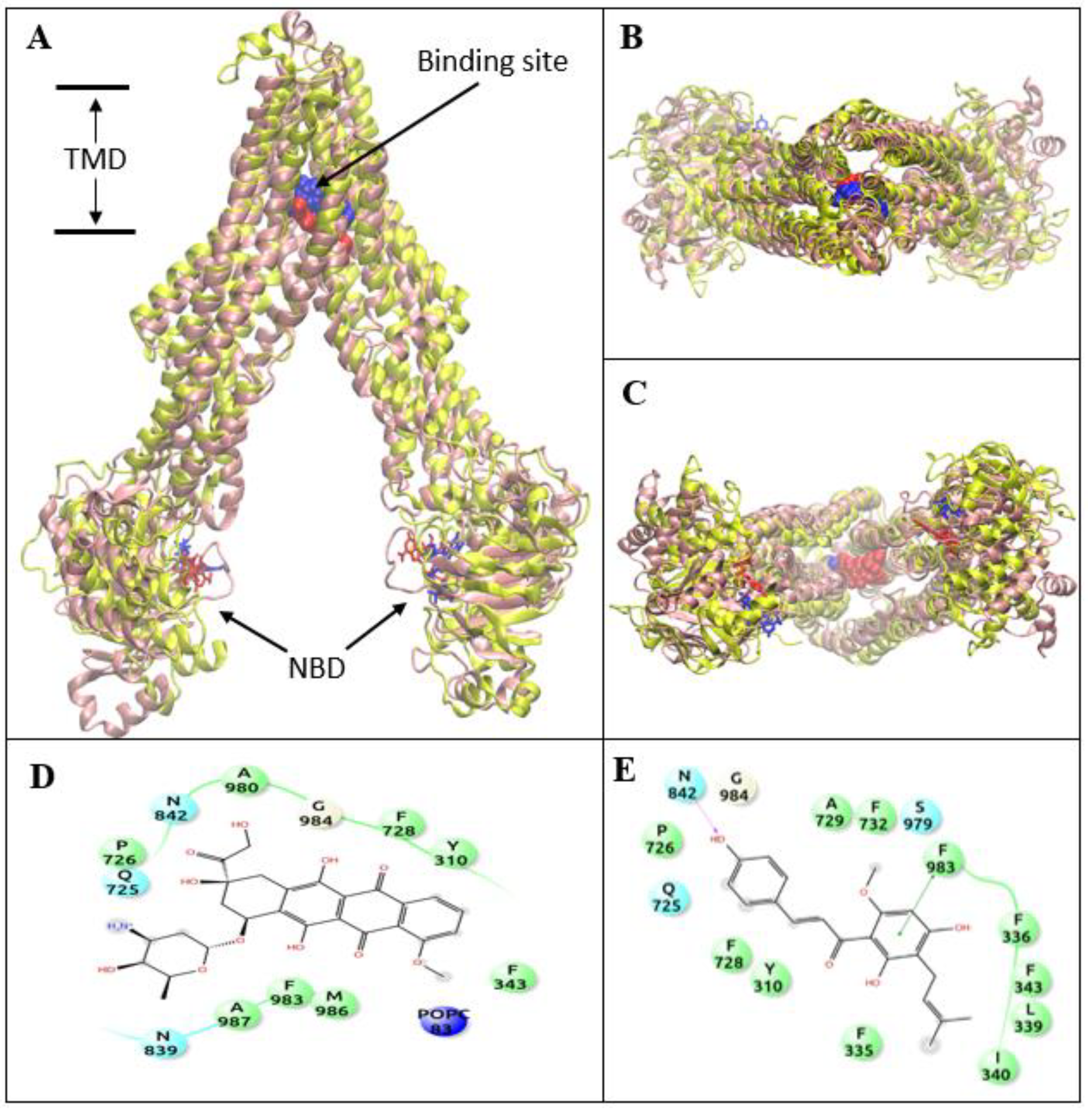
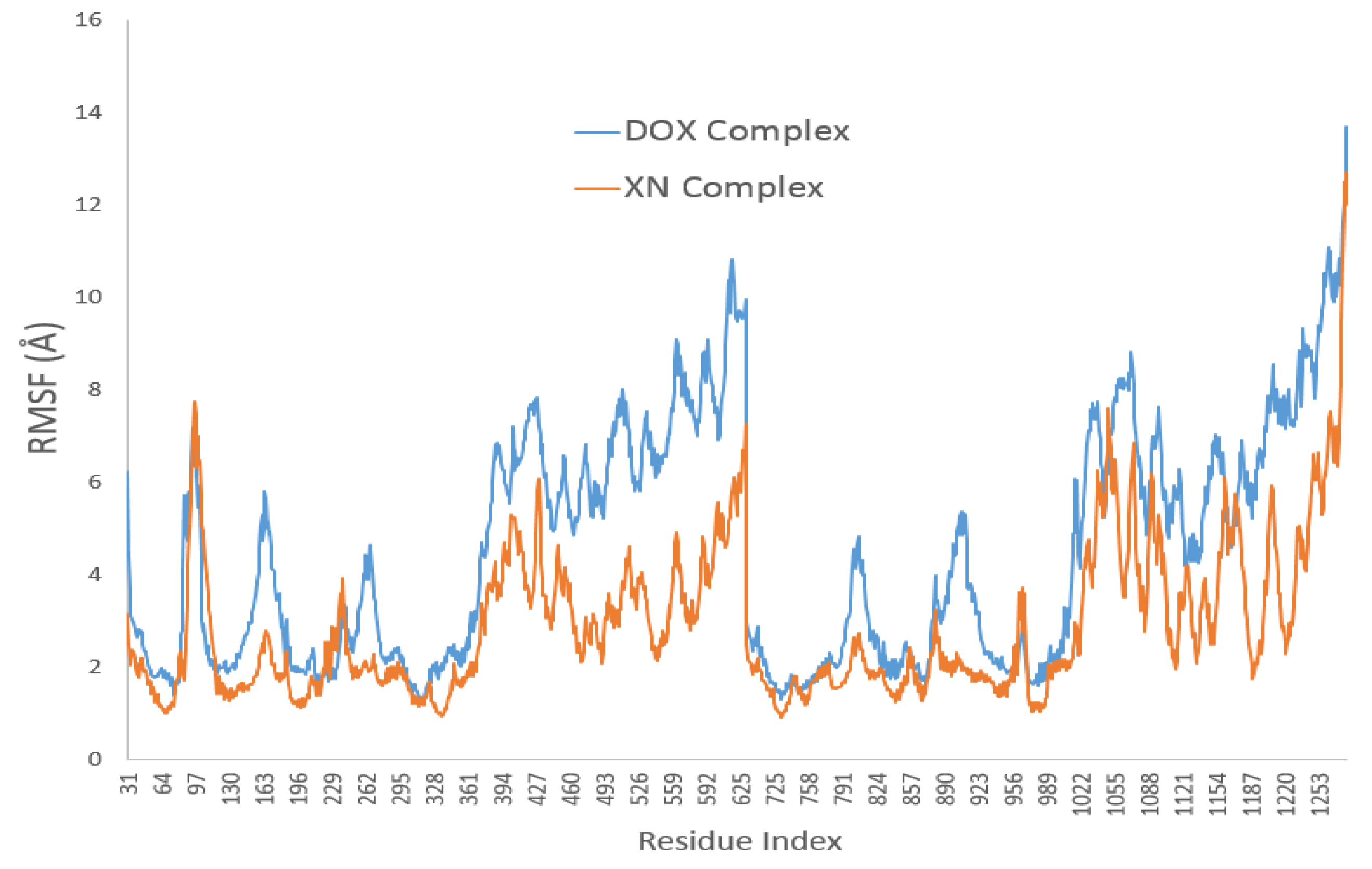
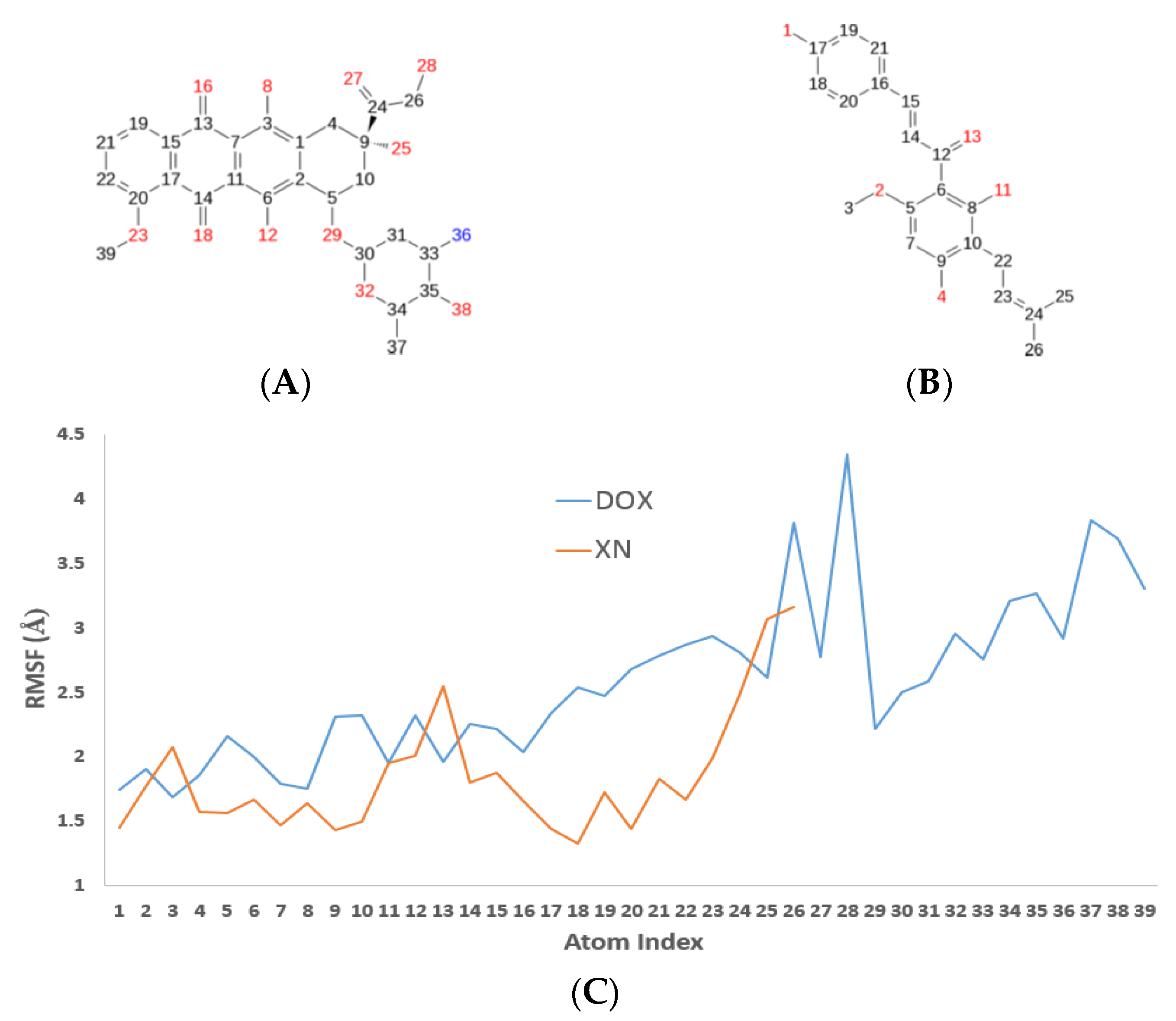
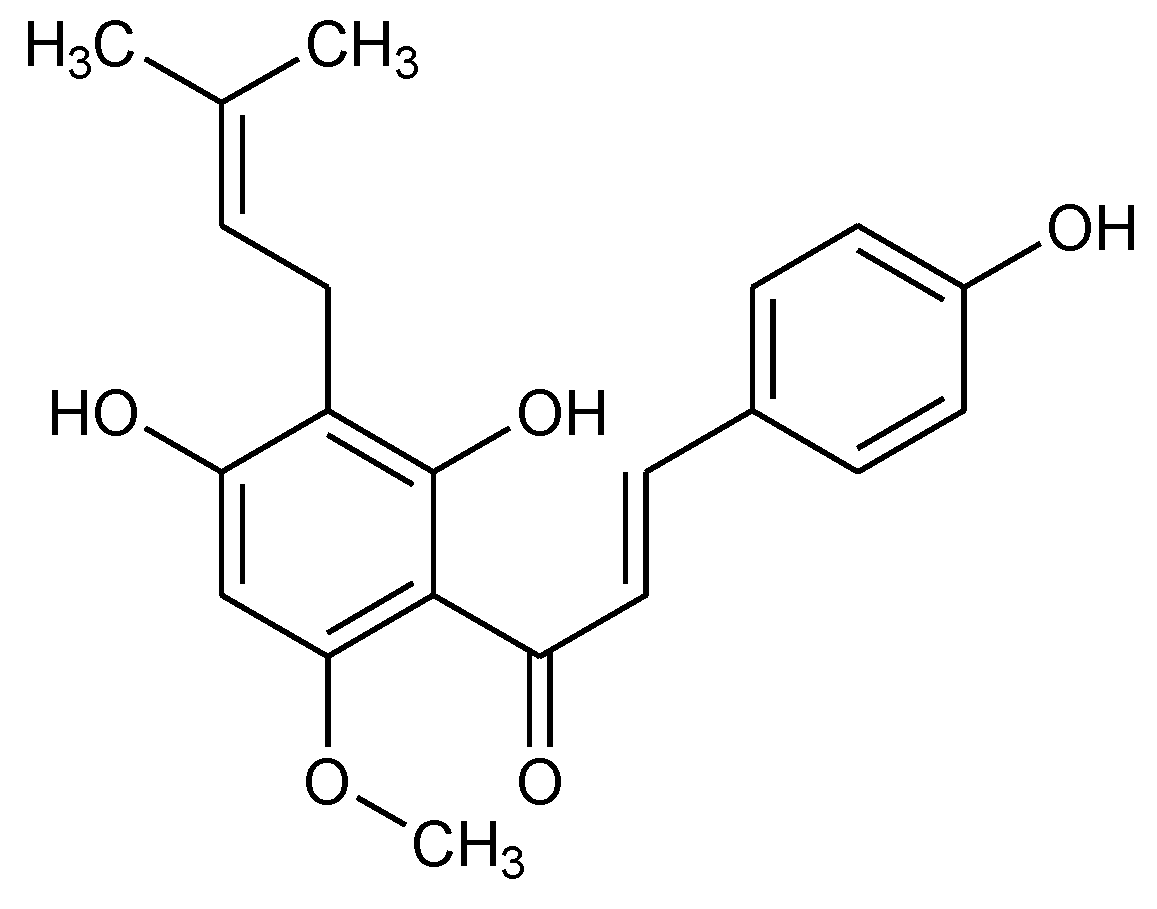
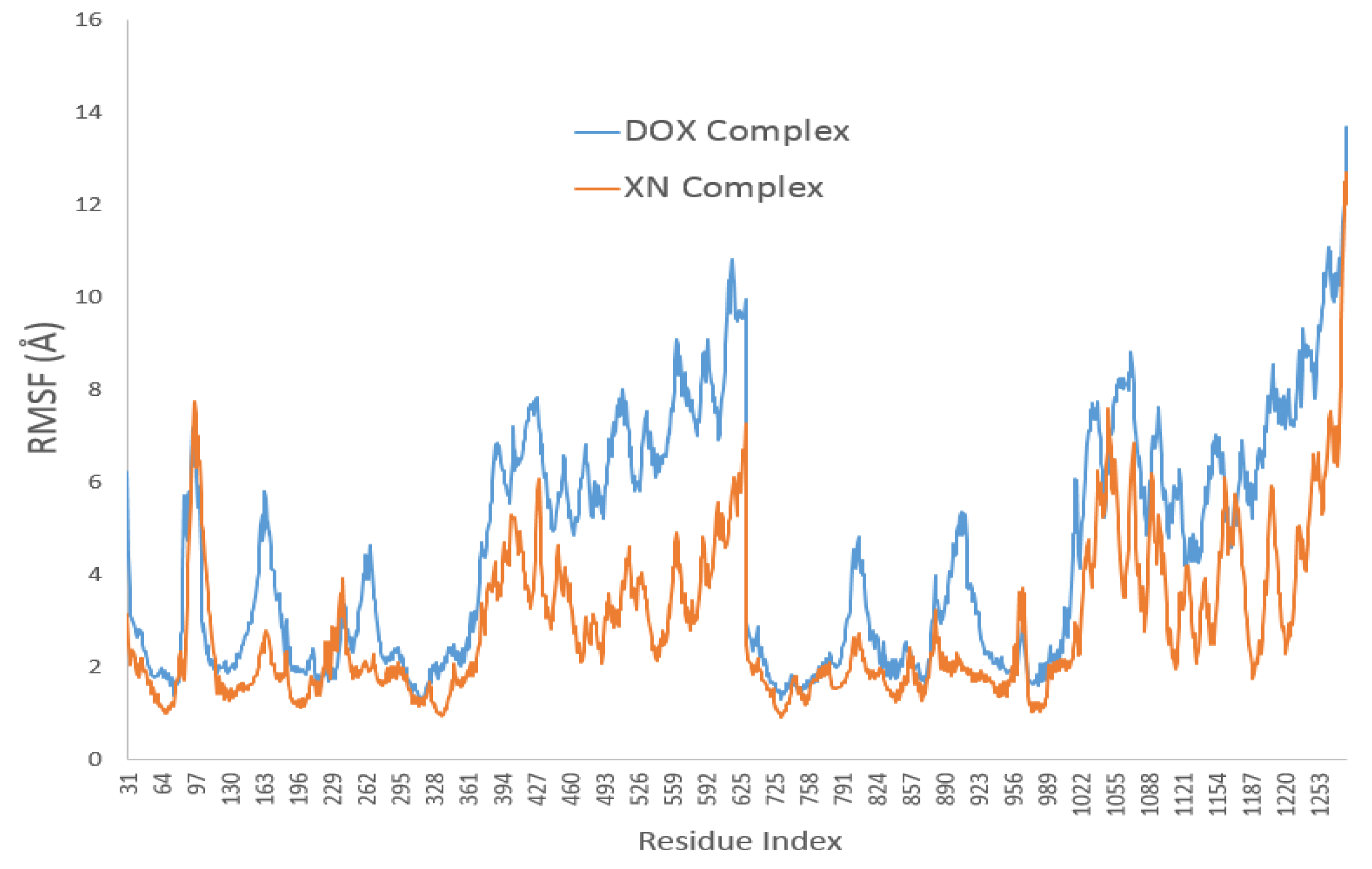
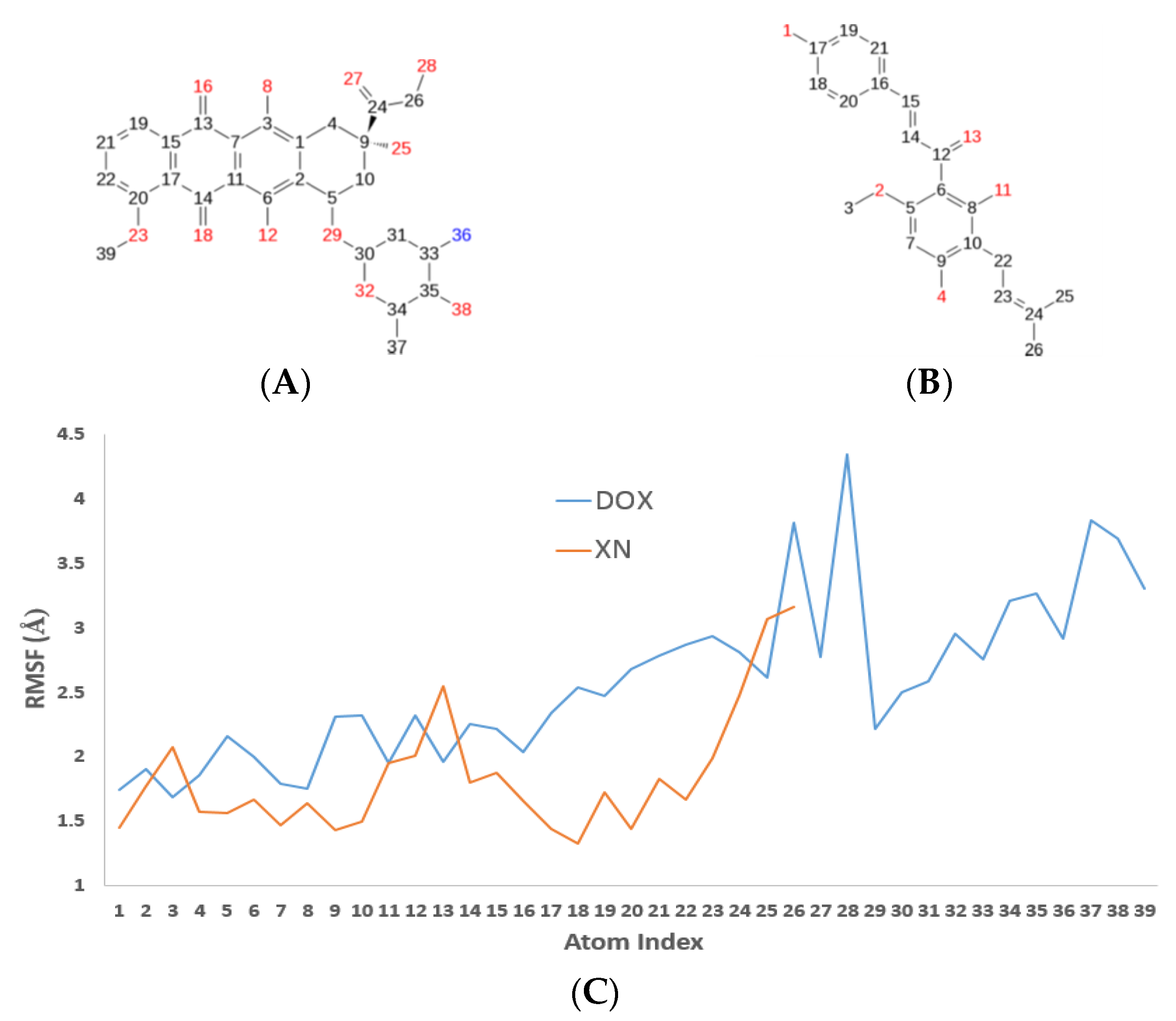
3.4. XN Bound to the Central Transmembrane Domain (TMD) Site, Overlapping with the DOX Binding Site
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Safar, Z.; Vasko, B.; Ritchie, T.K.; Imre, G.; Mogyorosi, K.; Erdo, F.; Rajnai, Z.; Fekete, Z.; Szeremy, P.; Muka, L.; et al. Investigating ABCB1-Mediated Drug-Drug Interactions: Considerations for In Vitro and In Vivo Assay Design. Curr. Drug Metab. 2016, 17, 430–455. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, T.; Efferth, T. P-glycoprotein and its inhibition in tumors by phytochemicals derived from Chinese herbs. J. Ethnopharmacol. 2012, 141, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hansen, P.E.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Bassani, B.; Camodeca, C.; Rosalia, L.; Cantelmo, A.; Gallo, C.; Baci, D.; Bruno, A.; Orlandini, E.; Nencetti, S.; et al. Synthesis and antiangiogenic activity study of new hop chalcone Xanthohumol analogues. Eur. J. Med. Chem. 2017, 138, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Dell’Eva, R.; Vene, R.; Ferrari, N.; Buhler, D.R.; Noonan, D.M.; Fassina, G. Mechanisms of the antiangiogenic activity by the hop flavonoid xanthohumol: NF-kappaB and Akt as targets. FASEB J. 2006, 20, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Park, M.A.; Heo, S.W.; Park, S.Y.; Kang, K.W.; Park, P.H.; Kim, J.A. The radio-sensitizing effect of xanthohumol is mediated by STAT3 and EGFR suppression in doxorubicin-resistant MCF-7 human breast cancer cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yin, H.; Qian, X.; Dong, J.; Qian, Z.; Miao, J. Xanthohumol, a prenylated chalcone from hops, inhibits the viability and stemness of doxorubicin-resistant MCF-7/ADR cells. Molecules 2016, 22, 36. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.W.; Cooney, J.; Jensen, D.; Li, Y.; Paxton, J.W.; Birch, N.P.; Scheepens, A. Hop-derived prenylflavonoids are substrates and inhibitors of the efflux transporter breast cancer resistance protein (BCRP/ABCG2). Mol. Nutr. Food Res. 2014, 58, 2099–2110. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, C.R.; Ivy, S.P.; Kao-Shan, C.S.; Whang-Peng, J.; Rosen, N.; Israel, M.A.; Melera, P.W.; Cowan, K.H.; Goldsmith, M.E. Isolation of amplified and overexpressed DNA sequences from adriamycin-resistant human breast cancer cells. Cancer Res. 1987, 47, 5141–5148. [Google Scholar] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Qi, C.; Sun, X.; Ma, X.; Zhang, H.; Hu, L.; Yuan, J.; Yu, Q. Arctigenin preferentially induces tumor cell death under glucose deprivation by inhibiting cellular energy metabolism. Biochem. Pharmacol. 2012, 84, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Ikeguchi, M. Partial rigid-body dynamics in NPT, NPAT and NP gamma T ensembles for proteins and membranes. J. Comput. Chem. 2004, 25, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.J.; Zhou, R.H.; Berne, B.J. Molecular dynamics with multiple time scales: The selection of efficient reference system propagators. J. Chem. Phys. 1996, 105, 1426–1436. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| XN (μM) | DOX (μM) | Effect | CI |
|---|---|---|---|
| 0.5 | 0.08 | 0.03 | 0.79 |
| 0.5 | 0.16 | 0.04 | 1.17 |
| 0.5 | 0.31 | 0.08 | 1.28 |
| 0.5 | 0.63 | 0.23 | 1.04 |
| 0.5 | 1.25 | 0.47 | 0.93 |
| 0.5 | 2.5 | 0.79 | 0.65 |
| 0.5 | 5.0 | 0.90 | 0.69 |
| 1 | 0.08 | 0.06 | 0.58 |
| 1 | 0.16 | 0.06 | 0.95 |
| 1 | 0.31 | 0.1 | 1.16 |
| 1 | 0.63 | 0.21 | 1.16 |
| 1 | 1.25 | 0.49 | 0.89 |
| 1 | 2.5 | 0.79 | 0.66 |
| 1 | 5.0 | 0.89 | 0.75 |
| 2 | 0.08 | 0.09 | 0.55 |
| 2 | 0.16 | 0.10 | 0.76 |
| 2 | 0.31 | 0.16 | 0.86 |
| 2 | 0.63 | 0.28 | 0.94 |
| 2 | 1.25 | 0.53 | 0.82 |
| 2 | 2.5 | 0.80 | 0.64 |
| 2 | 5.0 | 0.90 | 0.70 |
| ABCB1/Inhibitor Complex | ΔVDW | ΔGBELE | ΔLIPO | ΔTOT | ΔΔTOT |
|---|---|---|---|---|---|
| XN Complex | −51.6 ± 3.2 | 14.3 ± 3.2 | −50.7 ± 3.1 | −88.0 ± 6.3 | −1.9 |
| DOX Complex | −57.5 ± 3.4 | 11.4 ± 6.5 | −39.9 ± 3.0 | −86.1 ± 8.6 | 0.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Hoag, H.; Wu, C.; Liu, H.; Yin, H.; Dong, J.; Qian, Z.; Miao, F.; Liu, M.; Miao, J. Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1. Appl. Sci. 2018, 8, 681. https://doi.org/10.3390/app8050681
Liu F, Hoag H, Wu C, Liu H, Yin H, Dong J, Qian Z, Miao F, Liu M, Miao J. Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1. Applied Sciences. 2018; 8(5):681. https://doi.org/10.3390/app8050681
Chicago/Turabian StyleLiu, Fangming, Hannah Hoag, Chun Wu, Haizhou Liu, Hua Yin, Jianjun Dong, Zhonghua Qian, Feng Miao, Ming Liu, and Jinlai Miao. 2018. "Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1" Applied Sciences 8, no. 5: 681. https://doi.org/10.3390/app8050681
APA StyleLiu, F., Hoag, H., Wu, C., Liu, H., Yin, H., Dong, J., Qian, Z., Miao, F., Liu, M., & Miao, J. (2018). Experimental and Simulation Identification of Xanthohumol as an Inhibitor and Substrate of ABCB1. Applied Sciences, 8(5), 681. https://doi.org/10.3390/app8050681