1. Introduction
Over the past few years, conjugated oligomers and polymers have attracted considerable attention due to their applications in photovoltaic cells, light-emitting diodes (LEDs), field-effect transistors (FETs), electrochromic devices, chemical sensors, microelectronic actuators, etc. [
1,
2]. A large amount of experimental and computational work has been devoted to studying the electronic properties of conjugated polymers using density functional theory (DFT) by means of approximate values of energy and state density. However, this type of research is quite complicated because the polymers do not have a well-defined structure, many of them are insoluble and polydisperse, and there are far fewer computational techniques for infinite polymers than well-defined molecules.
In recent years, DFT has gradually replaced semiempirical methods, Hartree–Fock (HF) theory and perturbation theory in research on conjugated oligomers and polymers. Despite the many successes of DFT, HF theory gives a better approximation of the electronic properties of the CPs. To understand the behavior of the ground state of the electron and the interface of the material via photo-induced charge transfer, it is important to study CPs using DFT and nonadiabatic coupling calculations. To explore the electronic structure of the CPs, ab initio DFT molecular dynamic simulation can be studied in combination with quantum dynamic calculations of the electronic relaxation to investigate the electron transfer of CPs. For example, Han et al. [
3] studied the photo-induced charge transfer of an Au/Si metal-semiconductor nano-interface through a reduced density matrix (RDM), which was explained via Redfield theory and nonadiabatic coupling calculations based on the ab initio equations of a density function. The nonadiabatic couplings between electronic orbitals were processed on the fly along nuclear trajectories of the system [
3].
In this study, we considered a model of a polyacetylene (PA) which has no commercial use yet, although PA is used as a processing solution for film-forming conductive polymers and molecular electrons. PA polymers are one of the commonly used polymers for photovoltaics, LEDs (light-emitting diodes), diodes, and transistors; in these applications, one needs to design and operate materials with modified electronic properties [
3]. Therefore, electronic properties can be modified by using different types of approaches such as changing the composition of the polymer by adding doping, photoexcitation, or an injected charge, which occurs after applying different kinds of processes, such as photoexcitation, injection of charge, thermal motion (e.g., glass transition) or tight packing in the amorphous form of the material [
4]. During the last few decades, many researchers have worked on the electronic conductivity of polyacetylene, and it has received much attention among them, particularly because of the possibility of increasing its electronic conductivity by doping and the low dimensionality of the electrical conductivity [
5]. In 1977, for the first time, researchers published a study on the electrical conductivity of doped polyacetylene [
6]. Recently, Carter et al. [
7] explained the electronic and physical properties of trans-polyacetylene. In that article, they explained the infrared photoluminescence of PA which was used to study the electronic structure of PA. They concluded that the photogenerated excitons that were generated at or near
cis defects could become trapped at the
cis (—CH=CH—) bond and undergo radiative recombination [
7]. The observed photoluminescence emission energies were a function of the conjugation length of the
cis defect and the extent of local interchain π stacking. They proved that the
cis bond served to pinpoint and localize the photogenerated excitons formed on the polyacetylene chain [
7]. Analysis of the integrated photoluminescence intensities indicated that the percolation threshold for free carriers was achieved at just 30% overall
trans content, implying that thermally isomerized polyacetylene behaved more like a three-dimensional composite material than a one-dimensional material [
7]. Wong et al. [
8] explored the emission properties of
cis- and
trans-polyacetylene, which was controlled by the ratio of
cis- and
trans-polymers. They found that the emission efficiency and PL lifetime decreased with an increase in
cis content. Yoshino et al. explained the characterization of PL (photoluminescence) and EL (electroluminescence) by using polyacetylene derivatives. They experimentally proved that intensity of PL and the spectrum were dependent on the PA and various electrode configurations [
9]. Hidayat et al. studied the PL and EL of the mixtures of PA and found a remarkable spectral shift in PL and EL [
10]. Franco et al. [
11] studied the computational examination of
trans-polyacetylene oligomers, which was based on the electron vibrational dynamics of molecular systems. Flick et al. [
12] investigated the nonadiabatic contributions to the vibronic sidebands of equilibrium and explicitly time-resolved nonequilibrium photoelectron spectra for a vibronic model system of
trans-polyacetylene. They explained how the vibronic wavepacket motion of the model could be located in the time-resolved photoelectron spectra as a function of the pump–probe delay. Moliton et al. [
13] explained that after modification, π-conjugated polymers such as PA could be used for optoelectronics applications. Because of electronic correlation effects and strong electron–phonon interactions, the theoretical description of π-conjugated polymers is very complicated. From the previous studies on polyacetylene, it can be understood that [
14,
15] that coupling of the electrons to the nuclear degrees of freedom results depends on the semiconductor or crystalline metal materials of the PA. Tretiak et al. [
16] observed strong coupling to the nuclear degrees of freedom of the excited state electron nuclear dynamics of a polyacetylene chain. They explained the photoexcitation dynamics of conjugated
cis-polyacetylene oligomers using an ESMD quantum chemical approach, and found that the excitation moves to the lowest electronic energy and creates phonon excitations and significant local distortions towards the lattice. Gal et al. [
17] made a polymer using polyacetylene and experimentally proved that the polymer, which was prepared from polyacetylene, showed higher (352 nm) UV–visible absorption characteristics, and its highest photoluminescence spectrum was at 453 nm, which corresponds to the photon energy of 2.74 eV. Kim et al. [
18] reported the synthesis and properties of a new polyacetylene-based polyelectrolyte. The absorption spectrum exhibited a maximum absorption value of 480 nm, which was due to the p→p* transition of the conjugated polymer backbone. The photoluminescence spectra of the polymer exhibited a maximum peak of 550 nm, which corresponded to a photon energy of 2.26 eV. Typical irreversible electrochemical behaviors were observed between the doped and undoped peaks in the cyclic voltammograms of the polymer.
In this study, we have considered a simple single oligomer model of cis-polyacetylene to investigate interfacial electron transfer in the system, and representative nuclear configurations were obtained according to ab initio DFT molecular dynamic simulations and the time-dependent electronic structure. We considered the relaxation rates of photoexcited electrons and holes for our simulation using ab initio treatment of the electronic states when the underlying nonadiabatic transitions are accompanied by the respective vibrational dynamics. For this calculation, we used the Redfield theory for the calculation of the on-the-fly nonadiabatic couplings. which provide the dynamics of carriers and allow for an analysis of multiple nonradiative relaxation pathways in the cis-polyacetylene’s single oligomer. Photoexcitation can be found in this model by calculating the electron−phonon dynamics. In this work, we simulated our model using time-resolved emission spectra of the cis-polyacetylene, which provided some information about the lifetime of the emission state of the model. Our simulated results were only used for analyzing the thermal fluctuations of the lattice ions through nonadiabatic couplings, showing that for selected photoexcitations, the electron is promoted from carbon to hydrogen in the photon-mediated process and then recombines with the ground state calculation.
3. Results and Discussion
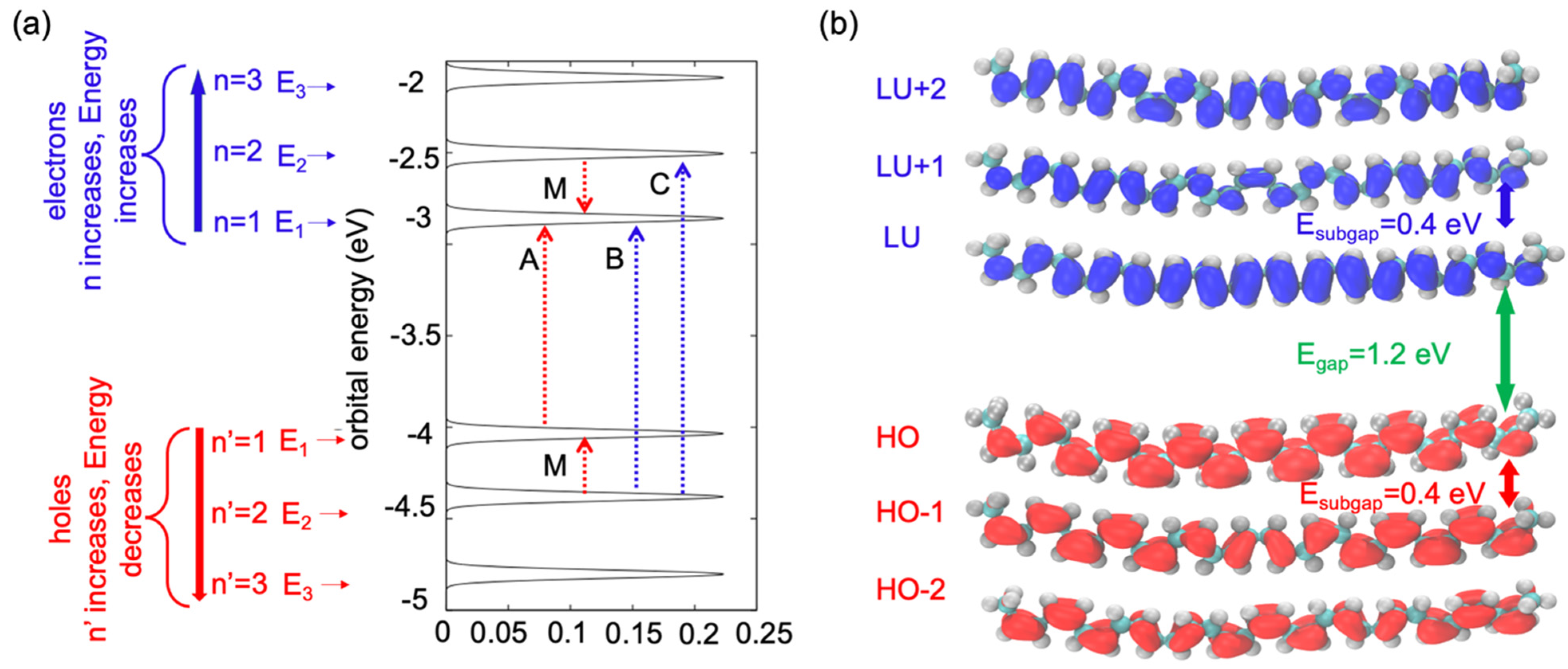
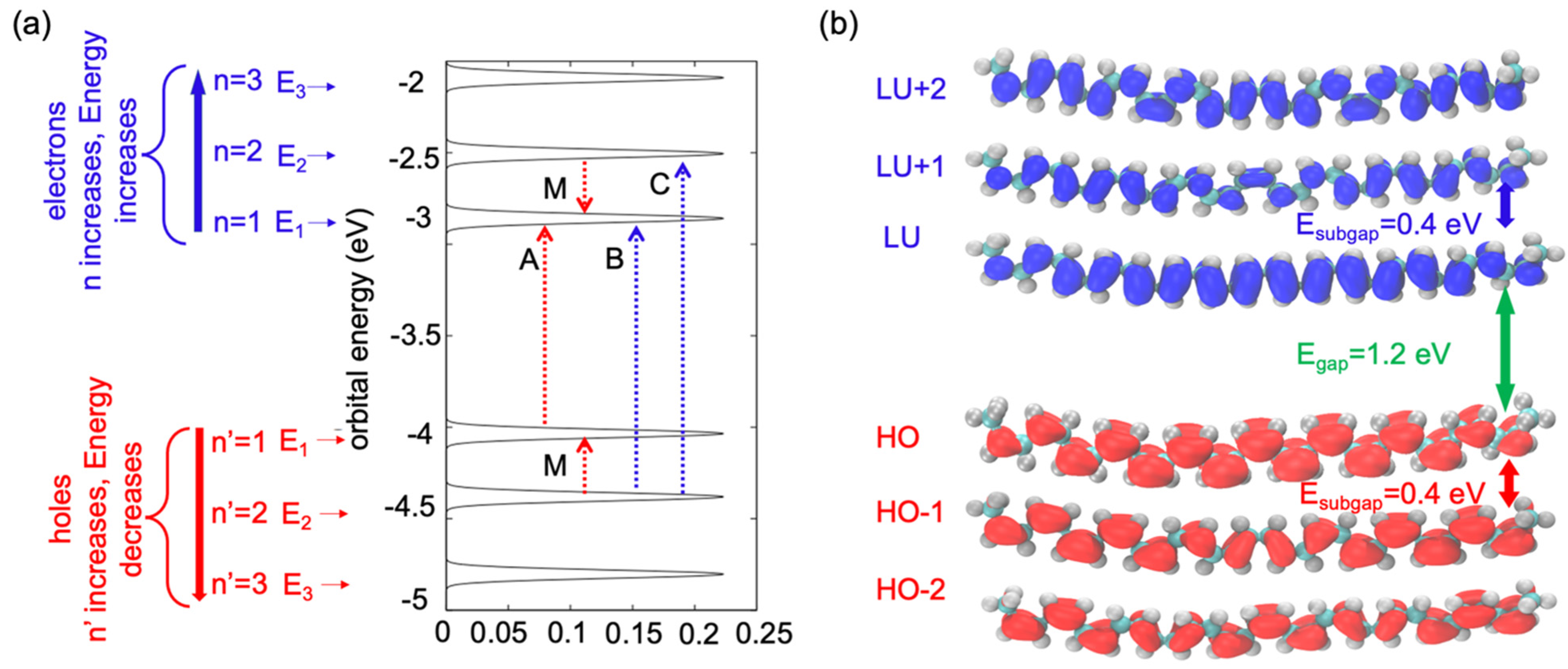
Figure 2 indicates the energy patterns of the frontier orbital. We used two labels for unoccupied and occupied orbitals: (a) each orbital was counted based on its occupation as
or
, and (b) through an analogy with the particle in the box, we labelled the unoccupied orbitals with the index
and the occupied orbitals with the index
. IN
Figure 2a, the LU orbital shows that the energy is
. The energies of the unoccupied orbitals increase with an increase in the number of indexes n. When
n = 2, the energy of the LU + 1 orbital is
and the sub-gap between the LU and LU + 1 orbitals is around 0.4 eV. However, the HO orbital shows lower energy (
). It was observed that the energy gap between the HO and LU was
, which was observed in the single-oligomer model. Because of the single oligomer model, there was less chance of hybridization between the electronic state of the orbital compared with a molecular crystal of multiple oligomers.
Figure 2a shows that the gap between the HO and LU orbitals was more than the sub-gap between the HO and HO + 1 orbitals, and between the LU and LU-1 orbitals.
Figure 2b illustrates the spatial isocontours of the frontier orbital
as computed by Equations (12) and (13). The maximum amount of the charge density of the LU orbital is shown in the middle portion of
, whereas a lower amount of the charge density appears at the edge of the orbital. The same trend is observed for the HO orbital: the maximum amount of the charge density appears in the middle portion of the particle in the box and a lower amount of charge density is shown at the edge of the particle in the box. When
n = 2 (the LU + 1 orbital and HO-1), the maximum amount of charge density changes from its original position. Because of the increase in the positive index of the eigenstate, the orbital was divided into two parts, and the maximum amount of the charge density was observed in the two middle portions of the orbital, and at the end of the edge and mid-point of the orbital, the charge density was almost zero.
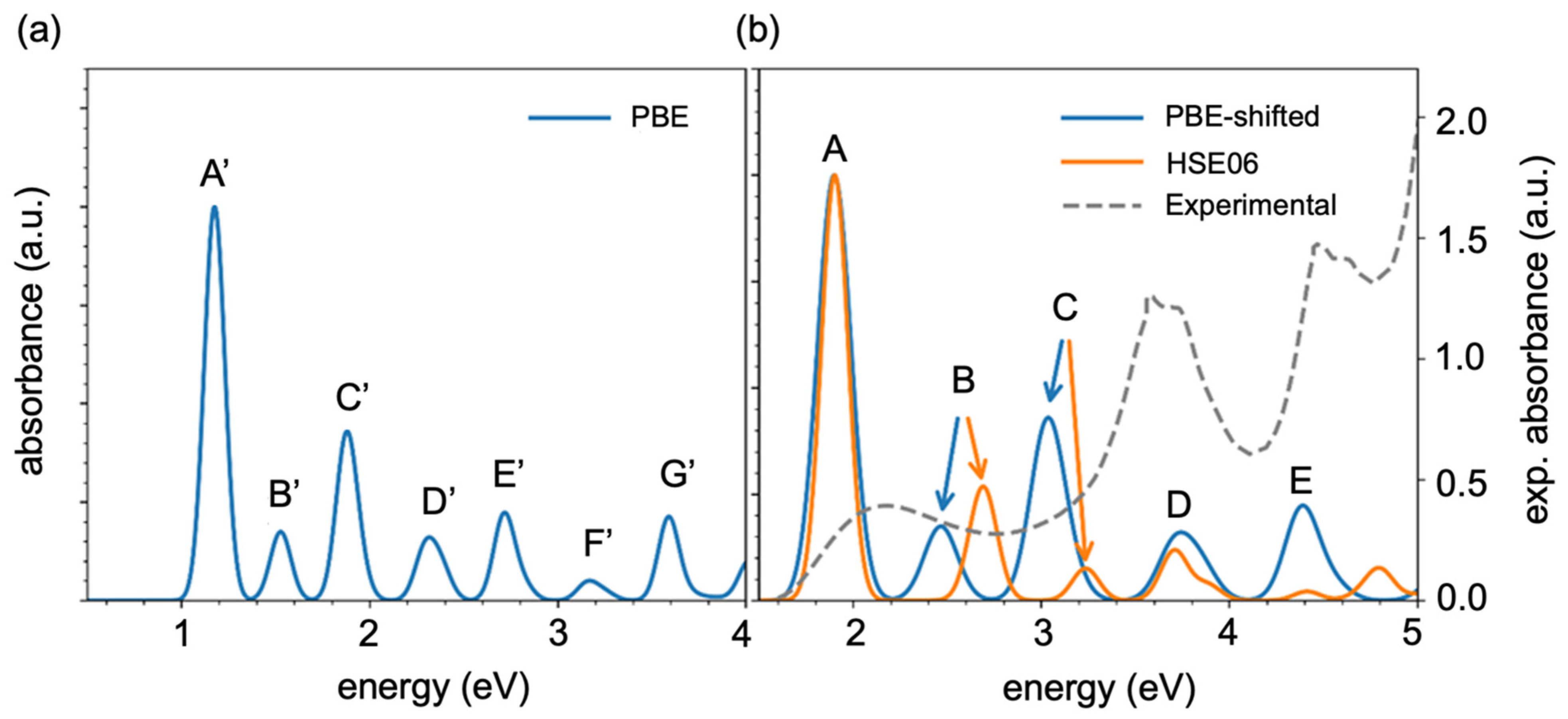
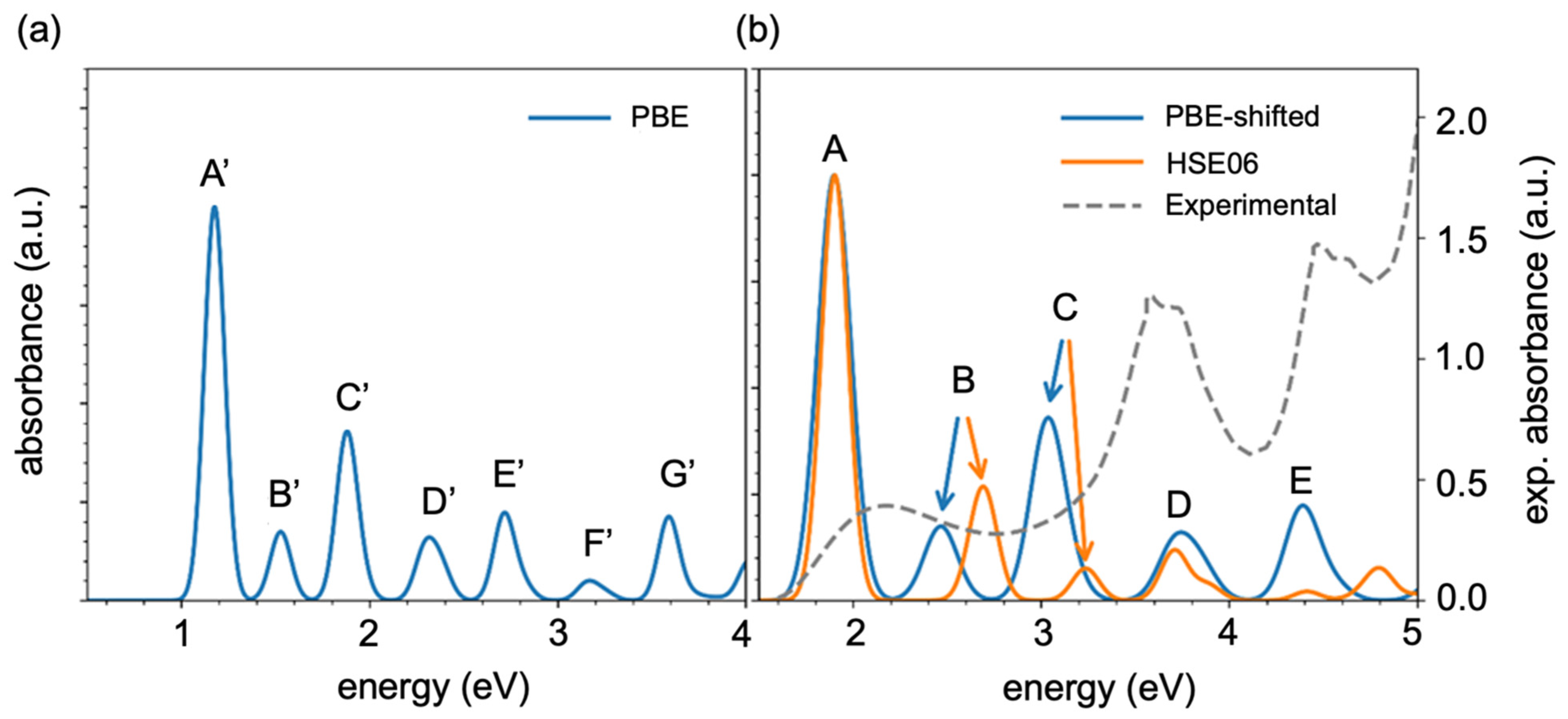
Figure 3a shows the absorption spectrum as a function of the transition energies computed by Equations (8)–(10), with intense peaks appearing from 1 eV to 4 eV. In
Figure 3a,b, the peaks are labeled in the order of ascending transition energy. Here, Peak A indicates the lowest transition energy but the highest intensity of absorption, which means that bright transition occurred, whereas Peak B, Peak D, and Peak F show the highest transition energies and lower intensities of absorption, which means that they show darker transitions compared with Peaks A’, C’ and G’. To validate this using the PBE GGA function, we simulated the absorption spectra by time-dependent DFT (TDDFT) (Equation (9)) theory with an HSE06 hybrid function using Equation (5). A comparison of the TDDFT simulated and experimental absorption spectra (
Table S1) indicates that the PBE GGA function underestimated the KS orbital energies, which demonstrates good qualitative agreement with the spectral features. [The comparison between the TDDFT simulated and experimental absorption spectra (
Table S1) indicates that the GGA functional PBE underestimated the KS orbital energies, which shows a good qualitative agreement with the spectral features] The absorption spectra can be calculated using the PBE function shifted by a ratio (R) of the bandgap in the HSE06 and PBE function calculations, R = E
gap(HSE06)/E
gap(PBE).
Figure 3b shows a comparison of the experimental spectra with the PBE simulated and HSE06 simulated absorption spectra, where the results obtained by the different functions (the PBE and HSE06 functions) qualitatively agreed with each other but there was a slight redshift in the B and C absorption peaks. Peak A shows the lowest transition energy but the highest intensity of absorption, and this occurred because of the bright transition. After we compared the simulated results with the experimental spectra, it can be concluded that the transition energies seem to agree, but the intensities and positions of peaks do not, probably because here, we looked at a single oligomer but, in the experiment, there was an ensemble.
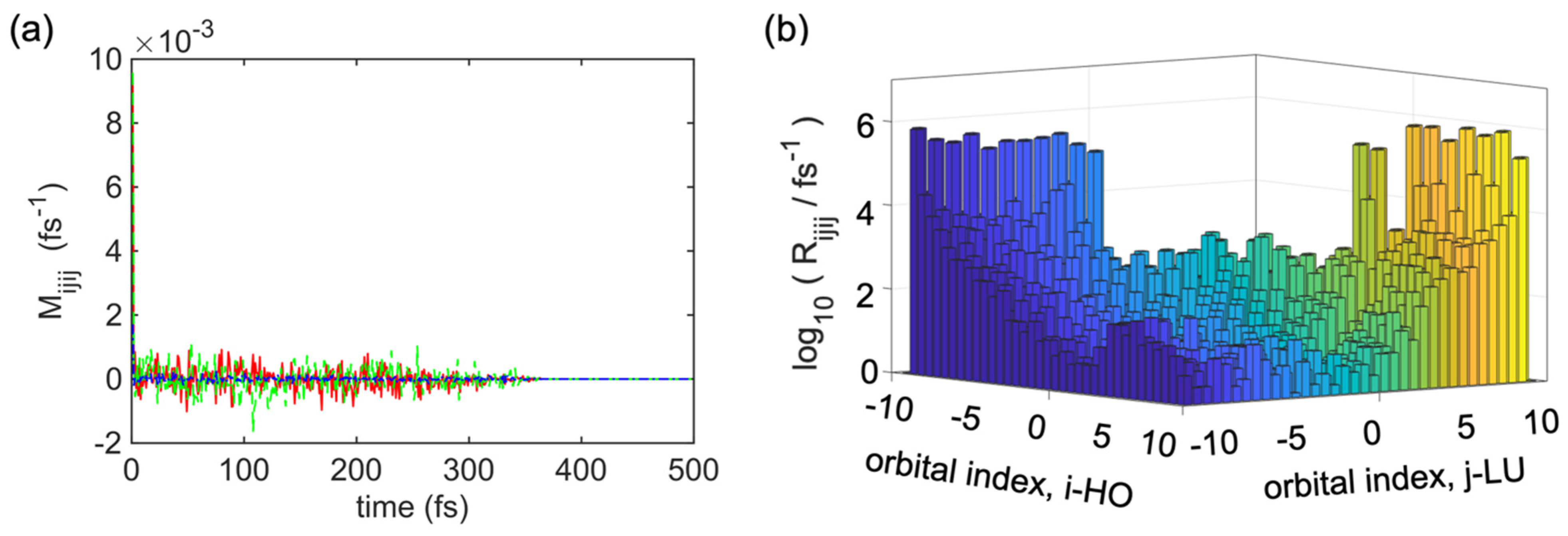
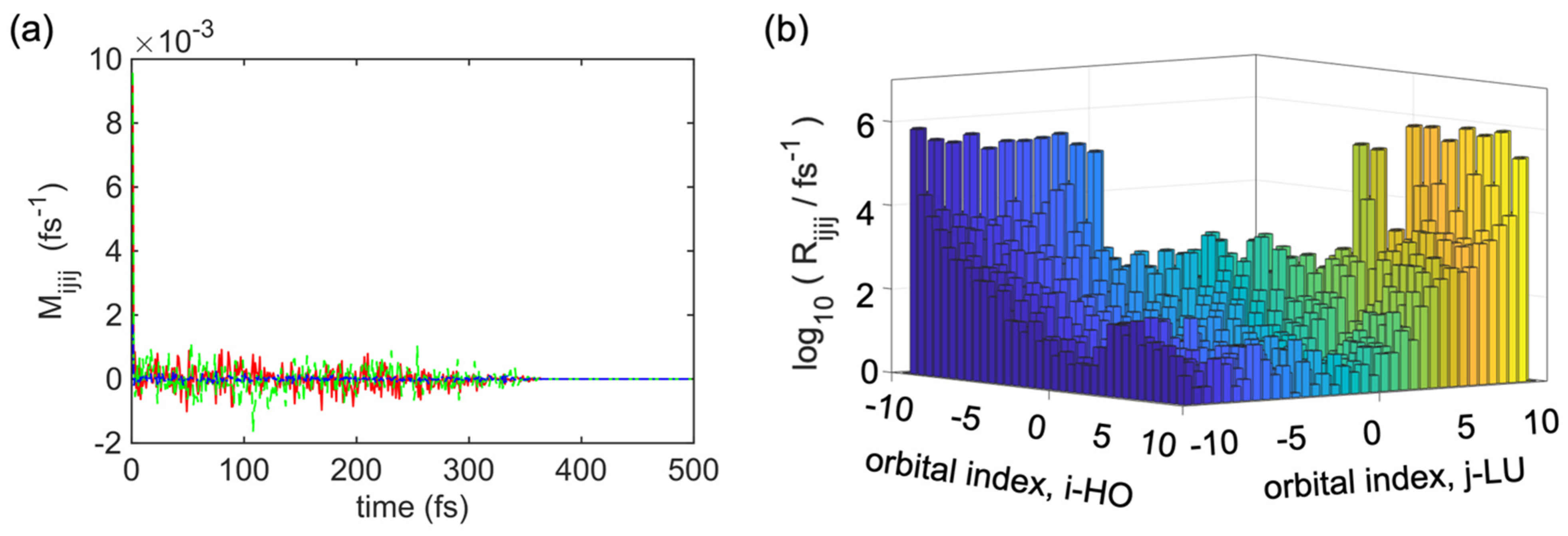
Figure 4a describes several examples of
. Using Equations (14)–(17), we can explain the average nonadiabatic interaction using the autocorrelation function of the electron-lattice interaction. This autocorrelation function shows several amplitudes, which provides information about the intensity of dissipative electronic transitions for the given indices
. Interestingly, for any tested combination of indices, the autocorrelation function decayed abruptly within less than 5 fs, and from 0 fs to 350 fs, it showed a fluctuation in the autocorrelation function of the electron-to-lattice nonadiabatic interaction (as explained in what follows), and after 350 fs, there was no fluctuation in the autocorrelation function, as indicated by the straight line which goes up to ∞ fs. This result justifies the Markovian approximation and time-independent form of the relaxation kernel in Equations (18) and (13) [
18].
At first, we extracted the Redfield tensor
from the nonadiabatic calculations of a single oligomer of the undoped
cis-polyacetylene then simulated the photoexcitation dynamics (time evolution of electron−hole pairs of orbitals) of the model.
Figure 4b shows the absolute values of selected elements of
, where the blue diagonal lines refer to the valence band (VB) and the yellow diagonal lines indicate the conduction band. There are several diagonal lines from the left bottom corner to the right top corner. In the middle section, some diagonal lines are smaller than others, and the mid-section shows the lowest transition rates, which refer to nonradiative transitions, most probably occur between the nearest neighboring orbitals
. Similar results for the dominant contribution of the phonon-mediated transitions between the nearest neighboring states were obtained for
.
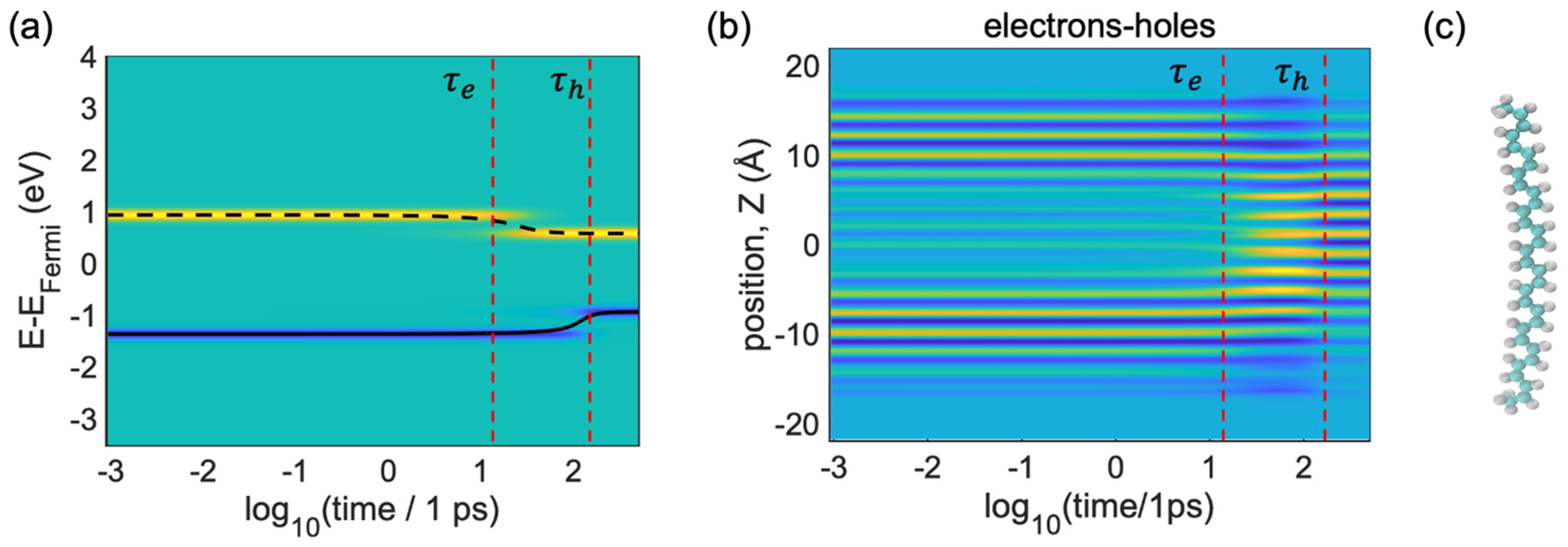
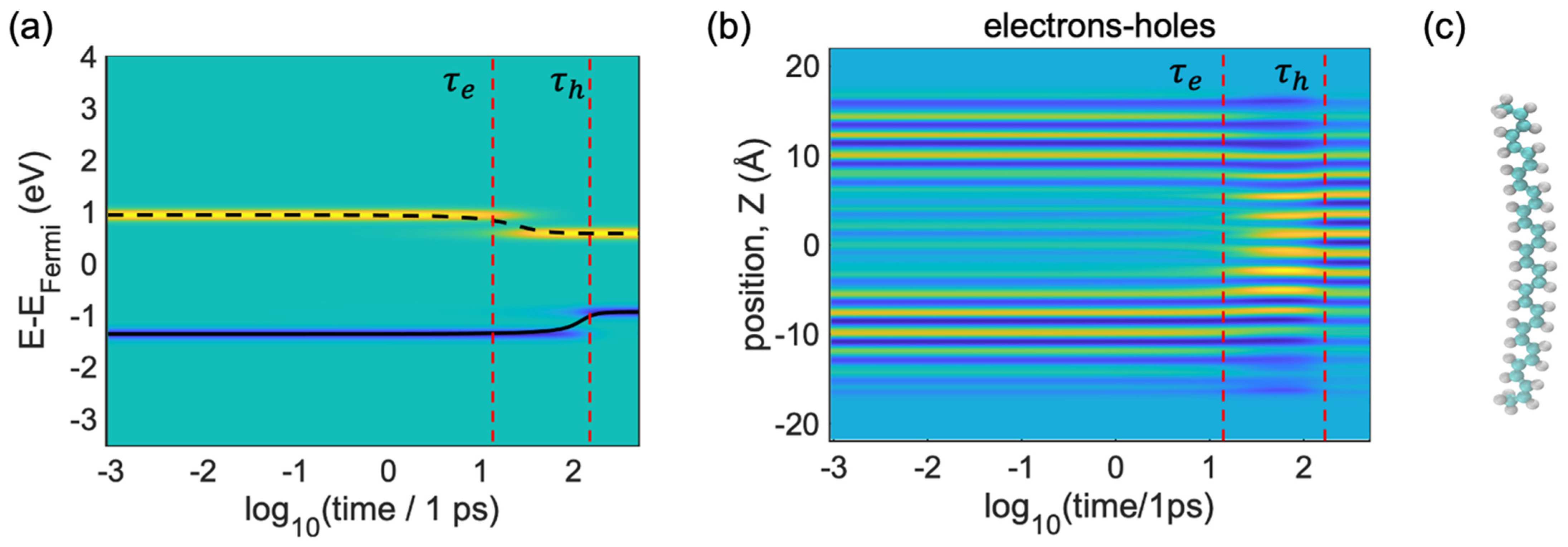
These simulation results shown in
Figure 5a summarize the evolution of electron and hole states, starting from the initial photoexcitation calculated by Equation (20) with
,
. The iso-contours of the population are shown in
Figure 5a, which can be
, as obtained from Equation (23), with yellow (blue) color indicating a large gain (loss) in charge density for the equilibrium distribution. In
Figure 5,
and
define the time of relaxation/cooling for the electron and the time of relaxation/cooling for the holes. The input orbital energies and their population dynamics were provided by the ab initio electronic dynamic calculations of the model shown in
Figure 5c.
Figure 5a illustrates that the nonradiative internal conversion of the photoexcited electron and hole is completed within
and
, respectively. From
Figure 5a we can see two spatial areas, which are highlighted by yellow and blue color. These color codes illustrate the distributions computed by Equation (23). Here, yellow areas refer to the photoexcitation of the electrons
and the blue areas indicate the photoexcitation of the holes
. Following the yellow areas, the energy of the electron decreases from 1 eV to 0.8 eV.
is the energy relaxation of the electron, which is visible at
. After
, the energy distribution of the electron and the energy distribution of the hole is almost constant. So, from
to
, there is energy relaxation from the higher energy level (the LU + 1 orbital) to the lower energy level (the LU orbital); here, the energy of the electron is dissipated into heat. After that, the electron population arrives at the LU orbital. That is why the energy of the electrons started to convert and, after
, the energy of the electrons showed almost constant energy.
Similarly, the blue line refers to the photoexcitation of the holes. After
, the energy of the holes (valence band) jumps from −1.5 eV to −0.8 eV. The hole population goes from the lower orbital (HO-1) to the higher orbital (HO), and after
(the time transfer of the holes =
), we can see a transition from the HO-1 orbital (−1.5 eV) to the HO orbital (−0.8 eV); after that, the energy of the holes or valence band (blue lines) shows almost constant values. Therefore, from
Figure 5a, we can say that electrons relax faster than holes.
In
Figure 5b, we further analyze the evolution of the nonequilibrium charge density distribution projected onto the z-direction, calculated through visualization of the KS orbitals from Equation (31).
Figure 5b can be explained with the help of
Figure 1. We can see that the energy of the population transfers from higher energy to lower states. The vertical line
shows the instant of time when the maximum amount of charge density of the electrons (yellow areas) experience transfer and the vertical line
shows the maximum amount of charge density of the holes (blue areas) experiencing transfer; one can interpret this in terms of the pattern of particles in a box for both the occupied and unoccupied orbitals. From
Figure 5b, we can see the one-dimensional orbital shapes of the HO-1, HO (blue spots), LU and LU + 1 orbitals in the z-direction (yellow spots). Because of the pattern of the HO-1 and LU + 1 orbitals (
Figure 1), there are no yellow lines available in the middle section and at the edge of the box (
to
). After
, the yellow lines have already condensed in the center. Though the yellow lines (electrons) have already condensed after
, the blue areas (holes) are still at the edges. We observed that at a time between
and
, the blue line
and yellow lines
stay in different areas of the space. Thus, from
Figure 5b, we can say that the holes relax from the edges to the center.
Concomitant to
Figure 5a, this picture shows that an electron and a hole reach the LU and HO orbitals localized mainly on the
. Notably, the electrons’ nonradiative relaxation is faster
compared with the holes’ relaxation, possibly because of two reasons: (i) the energy of the excited hole’s orbital stays far behind the band edge, whereas the excited electronic orbital stays at the edge of the band, and (ii) the electron−phonon couplings are stronger than hole−phonon couplings.
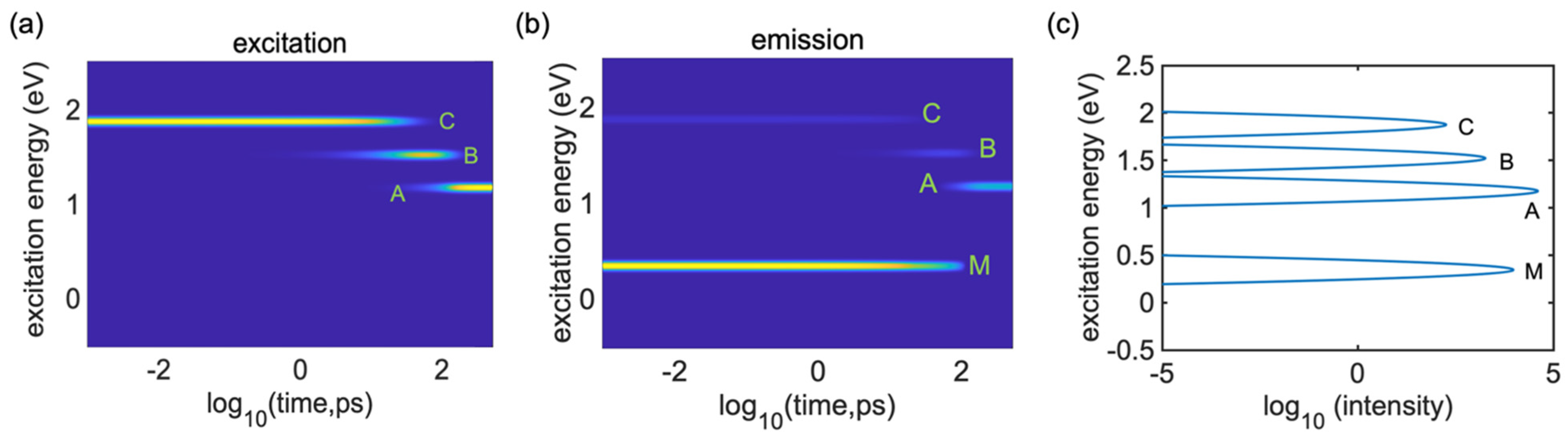
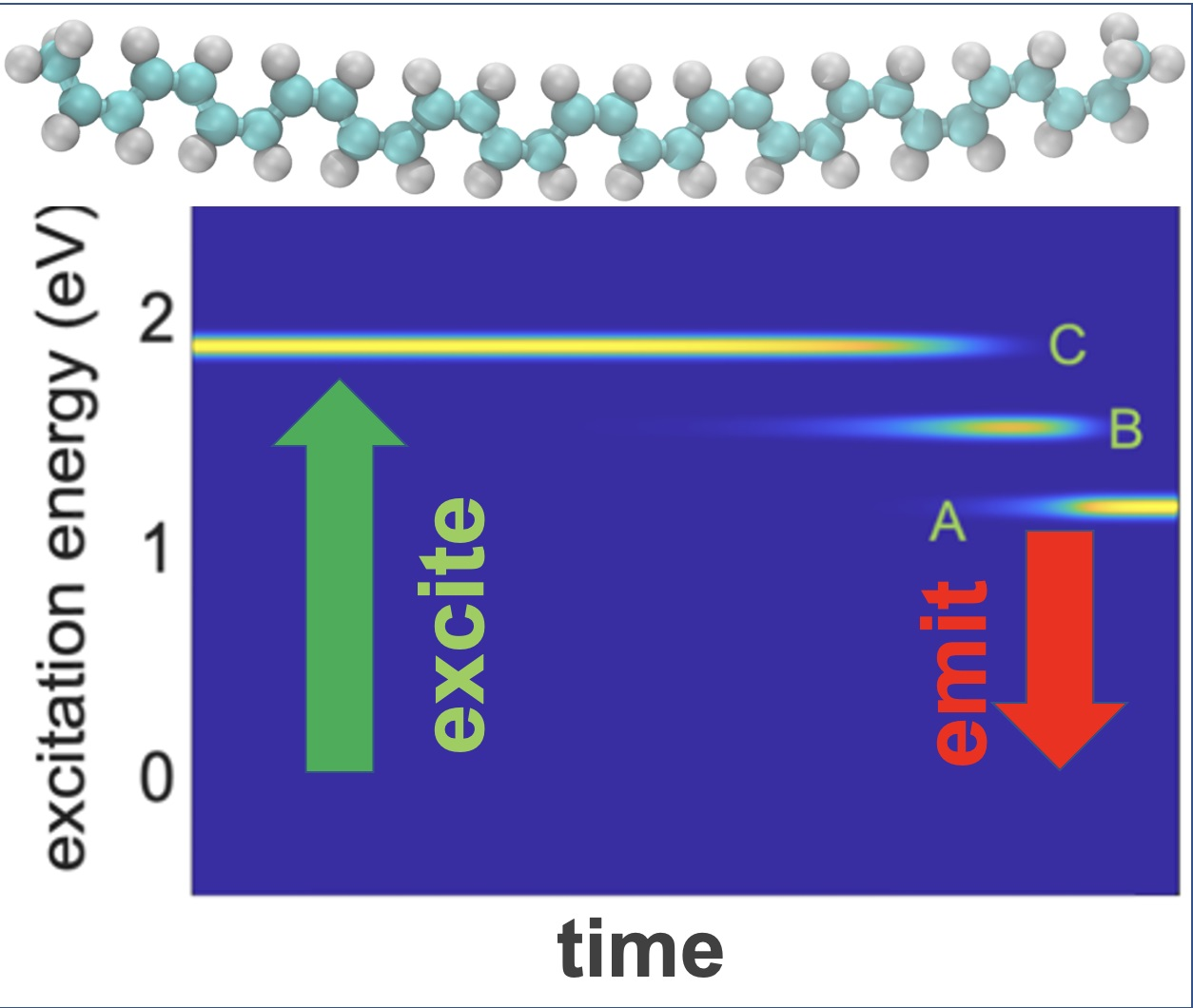
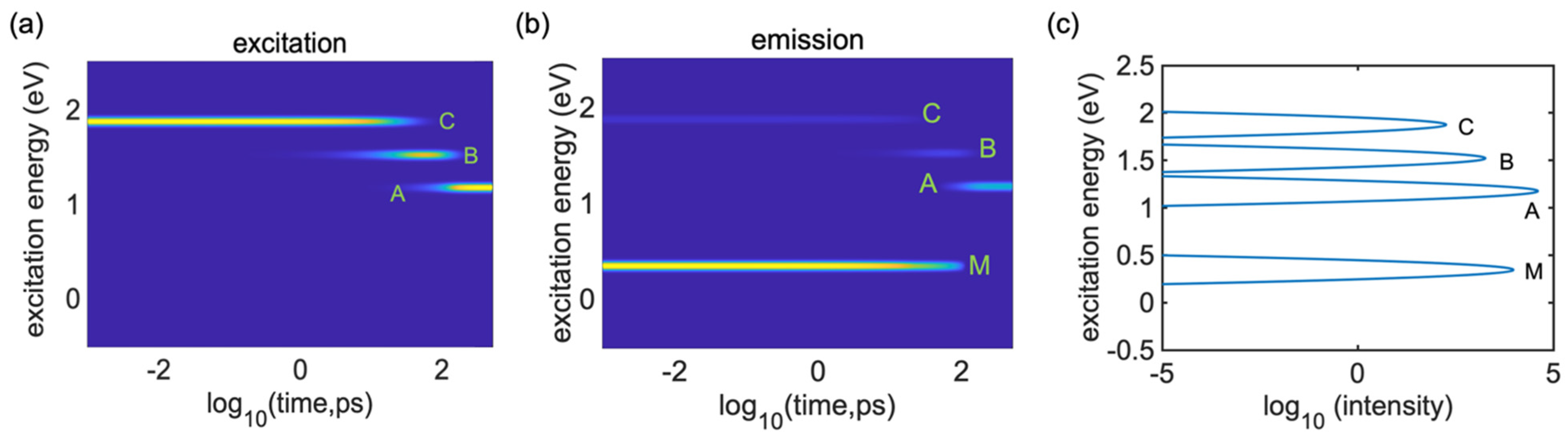
To study the dynamics of radiative and nonradiative energy dissipation in detail, we focused on photoexcitation with the energy of 2.0 eV promoting an electron from HO-1 to LU + 1, which corresponded to the D peak in the absorbance spectra.
Figure 6a clearly shows the electronic energy dissipation via the lattice vibrations which occurred between about
and
. Using Equation (33), we computed the time-resolved emission spectrum following the instantaneous photoexcitation at a transition energy of 2.0 eV, as displayed in
Figure 6b. An emission signal in the range of 2.0 eV, which corresponds to the parent inter-band absorption from the valence band (VB) to the conduction band (CB), disappeared within 0.1 ps, reflecting the beginning of the vibrational relaxation. In
Figure 6a, we can see the transition energies (Peak A, Peak B and Peak C) from 1.0 eV to 2.0 eV, which took
to
. Peak C showed higher photoluminescence; after that, it showed weaker photoluminescence, as seen from
Figure 6a (Peak B and Peak A). If we compare
Figure 6a with
Figure 6b, we cannot see any photoluminescence, which indicates very weak transitions. Due to the relaxation of the excitation energy, the energy started to disappear, showing the dark transition state of the system. Interestingly, there is one extra line that is visible at 0.2 eV in
Figure 6b, which shows the intra-band emission features at energies below the bandgap. This also appeared in the integrated emission spectrum graph (Peak
) but this peak is not observable in the absorption spectrum (
Figure 3a) and it is noticeable in the integrated emission spectrum graph.
Figure 6c presents the respective integrated emission spectrum of the single undoped
cis-polyacetylene oligomer after 2.0 eV photoexcitation, which was computed by Equation (34) and demonstrates that the emission peaks are consistent with the features in
Figure 6b. However, the emission features around B and C are very weak and are barely noticeable in
Figure 6b, but these peaks (Peak B and Peak C) emerge in the integrated emission plot. These excitations are activated in the later stages of the dynamics (after
) and stay active for a longer period (up to
), providing a noticeable contribution to the integrated emission Peaks C at 2 eV, B at 1.5 eV and A at 1 eV.
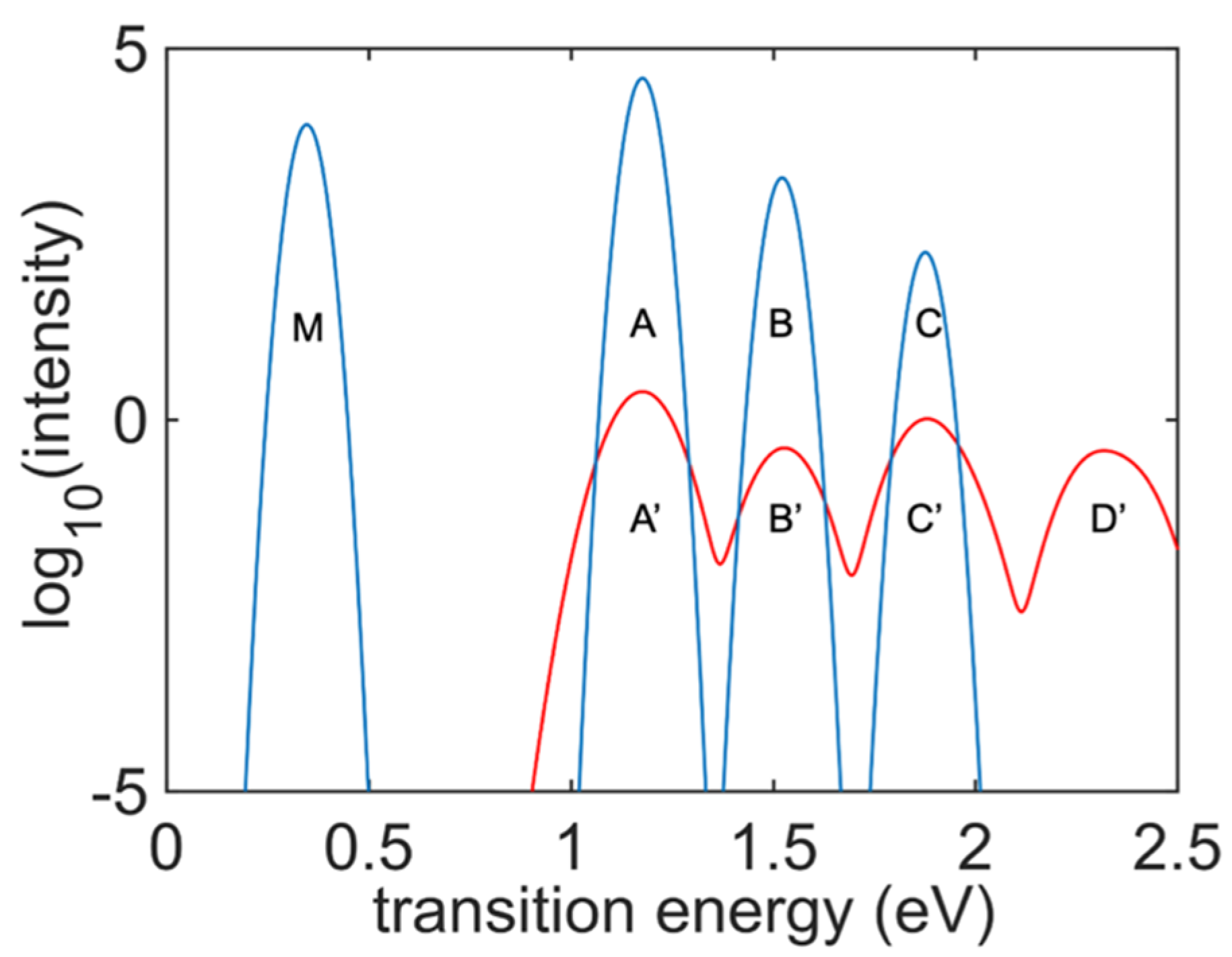
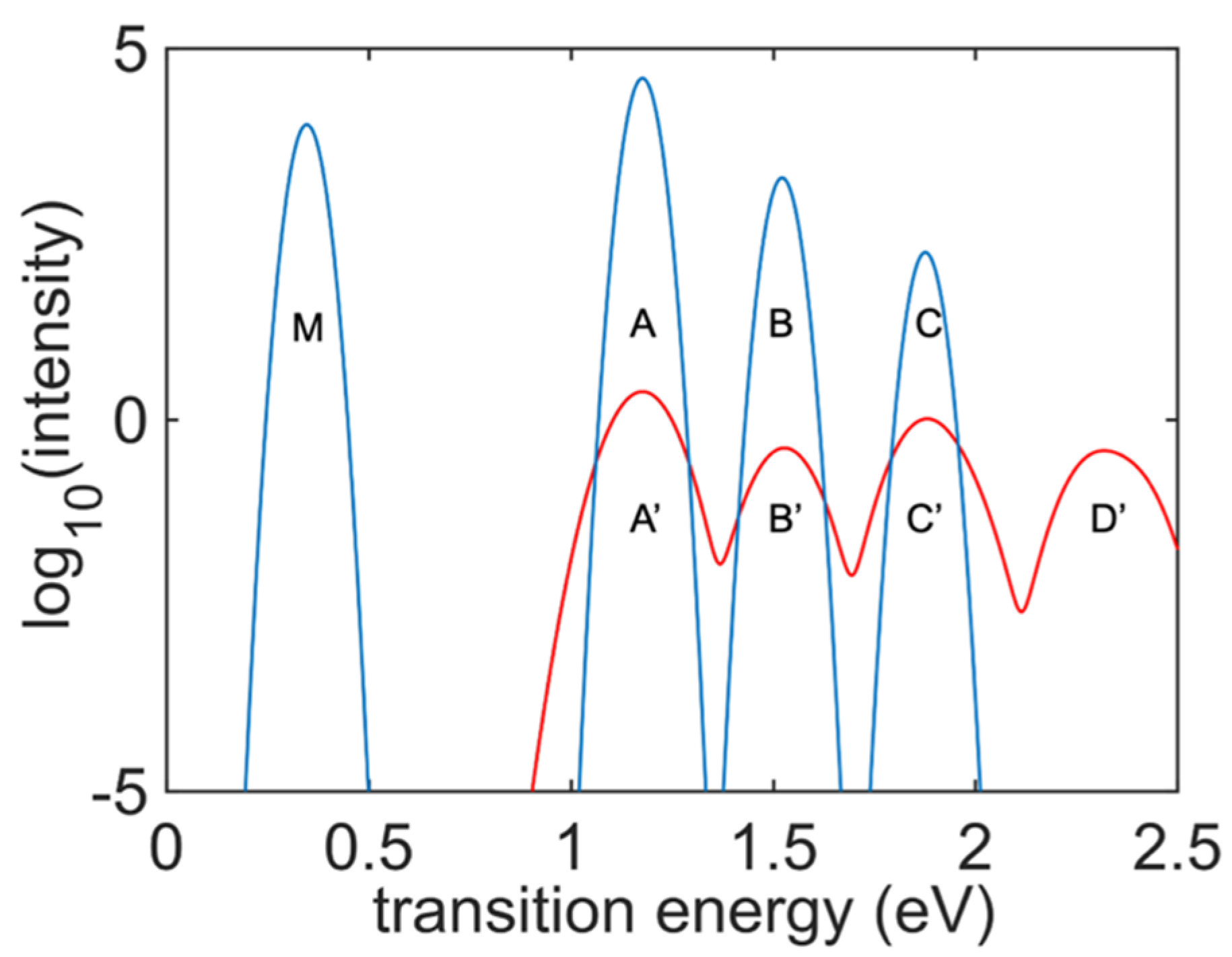
Figure 7 presents a comparison of the absorption and emission spectra of the undoped
cis-polyacetylene oligomer. The absorption spectrum almost overlaps with the respective integrated emission spectrum of the model. Peak
′ shows the lowest inter-band transition energy, which is also visible in the integrated emission spectrum. All transition energies are weaker than the other excitation energies, and one transition energy peak
′ is absent in the integrated excitation energy graph since excitation for the PL was performed at the transition energy of Peak C. There is one additional peak (the blue Peak
) that is visible only in the integrated emission spectrum graph but is missing in the absorption spectrum. This peak originates from transitions in the IR range, when an electron and a hole experience transitions inside the bands. Such inter-band transitions are disabled in the ground state but become available in the nonequilibrium excited state when two orbitals belonging to the same band satisfy the inverse population criterion. Computational identification of the mechanism of intra-band emission is the main finding of this work.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}