
Chiral Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides—Their Biological Activity, Lipophilicity, Protein Affinity, and Metabolic Transformations

 ,
,  , ,
, , 

Abstract
1. Introduction
2. Results and Discussion
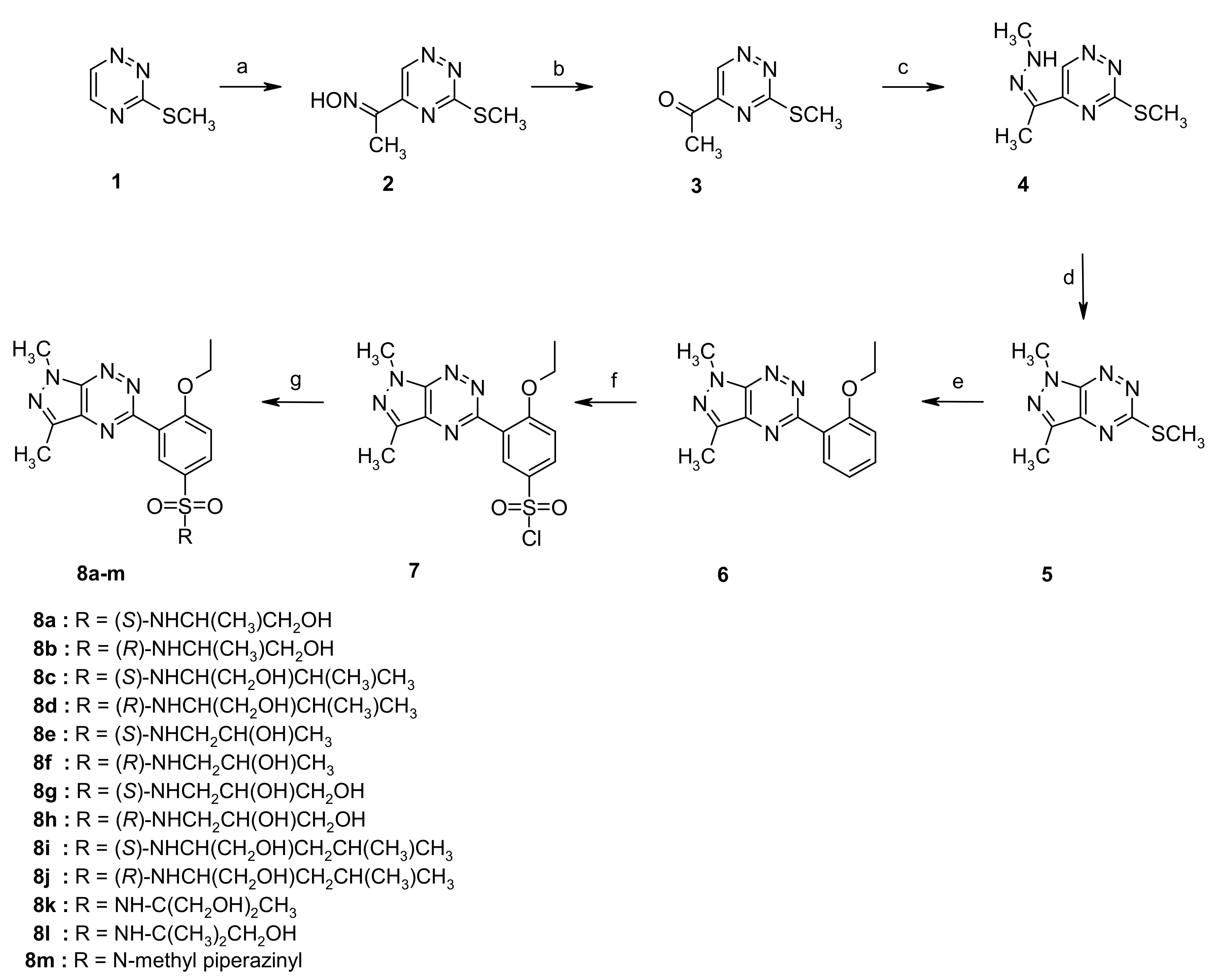
2.1. Chemistry
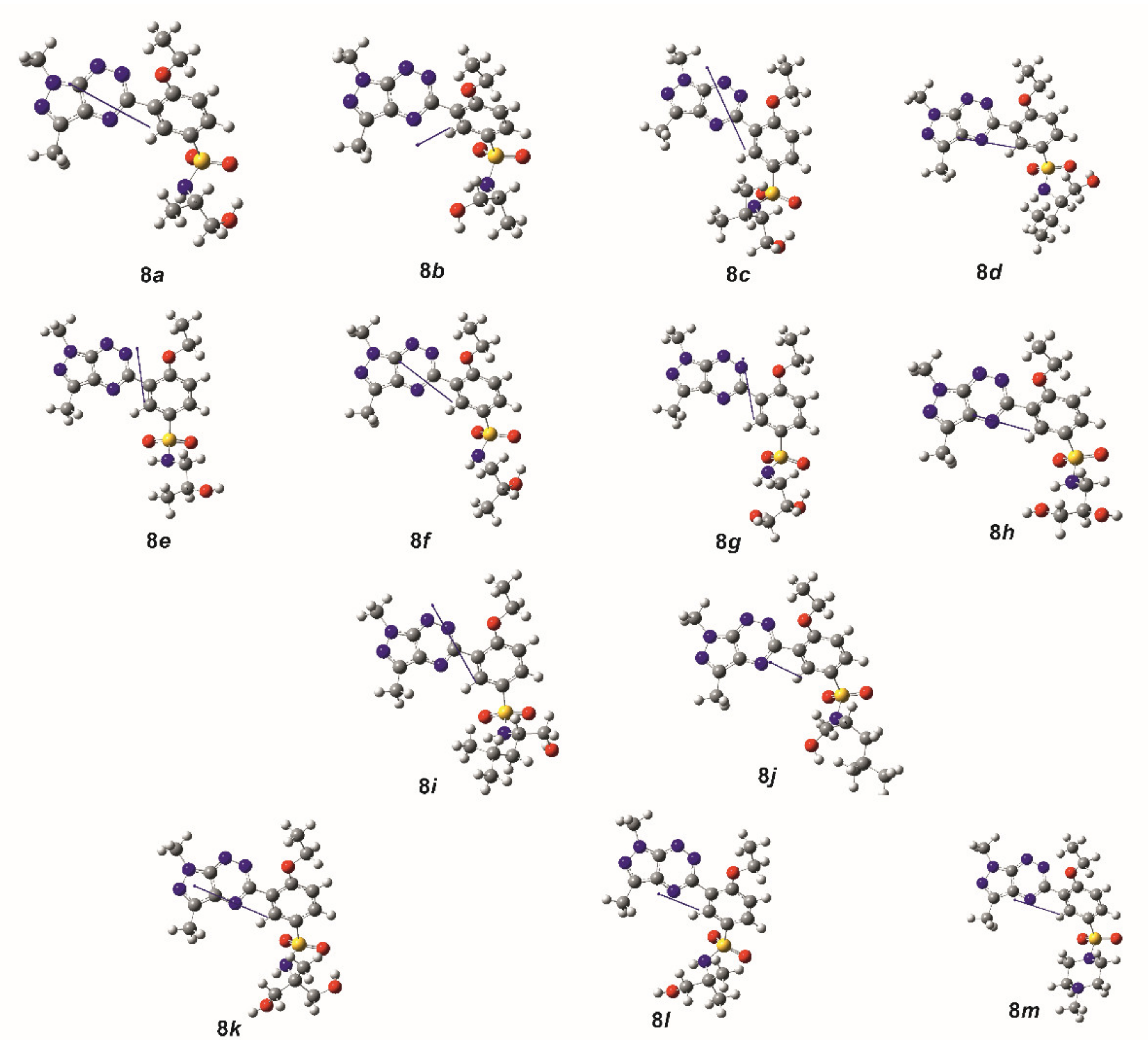
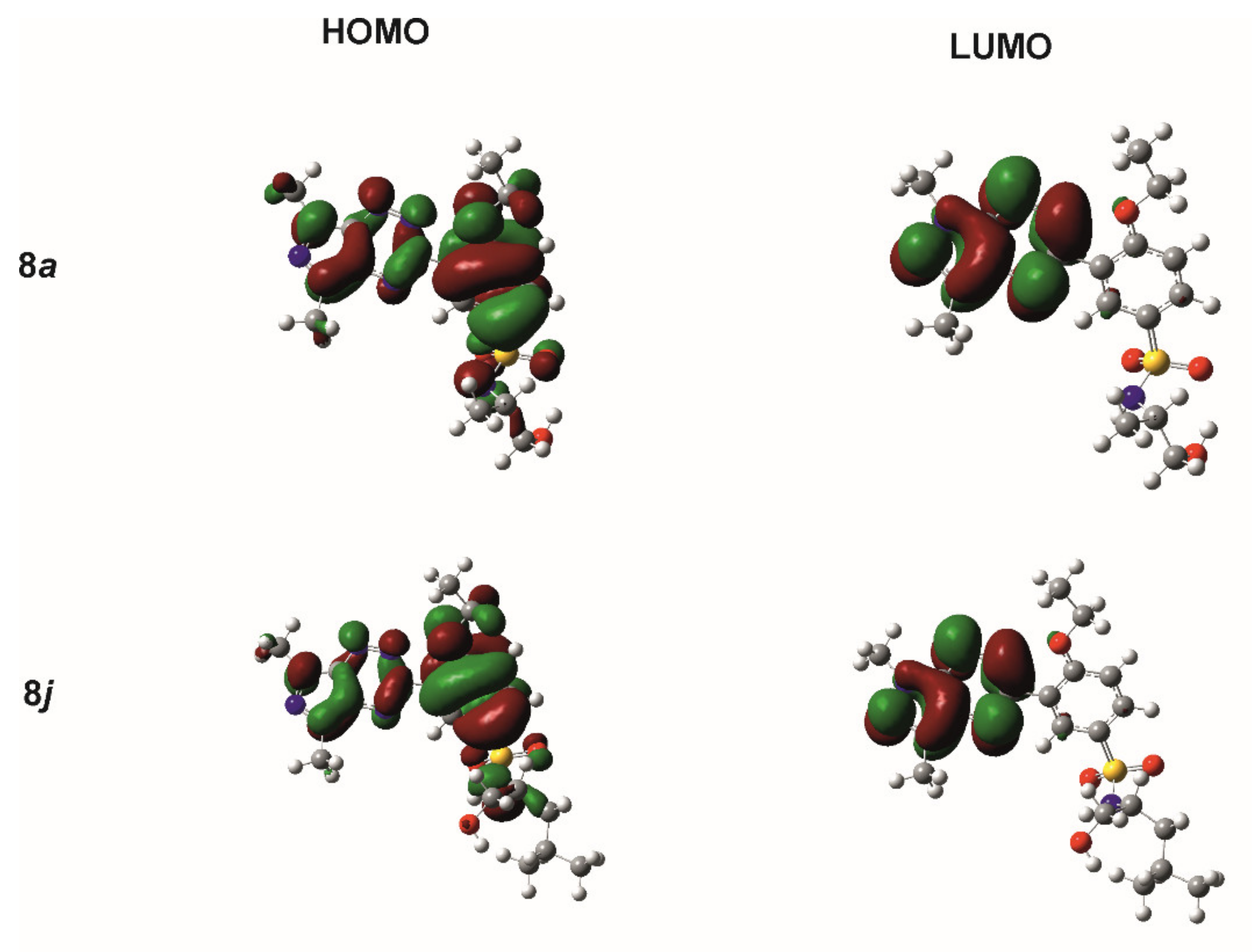
2.2. Theoretical Calculations
2.3. Antiproliferative Activity against Tumor Cell Lines
2.4. Lipophilicity and Protein Affinity
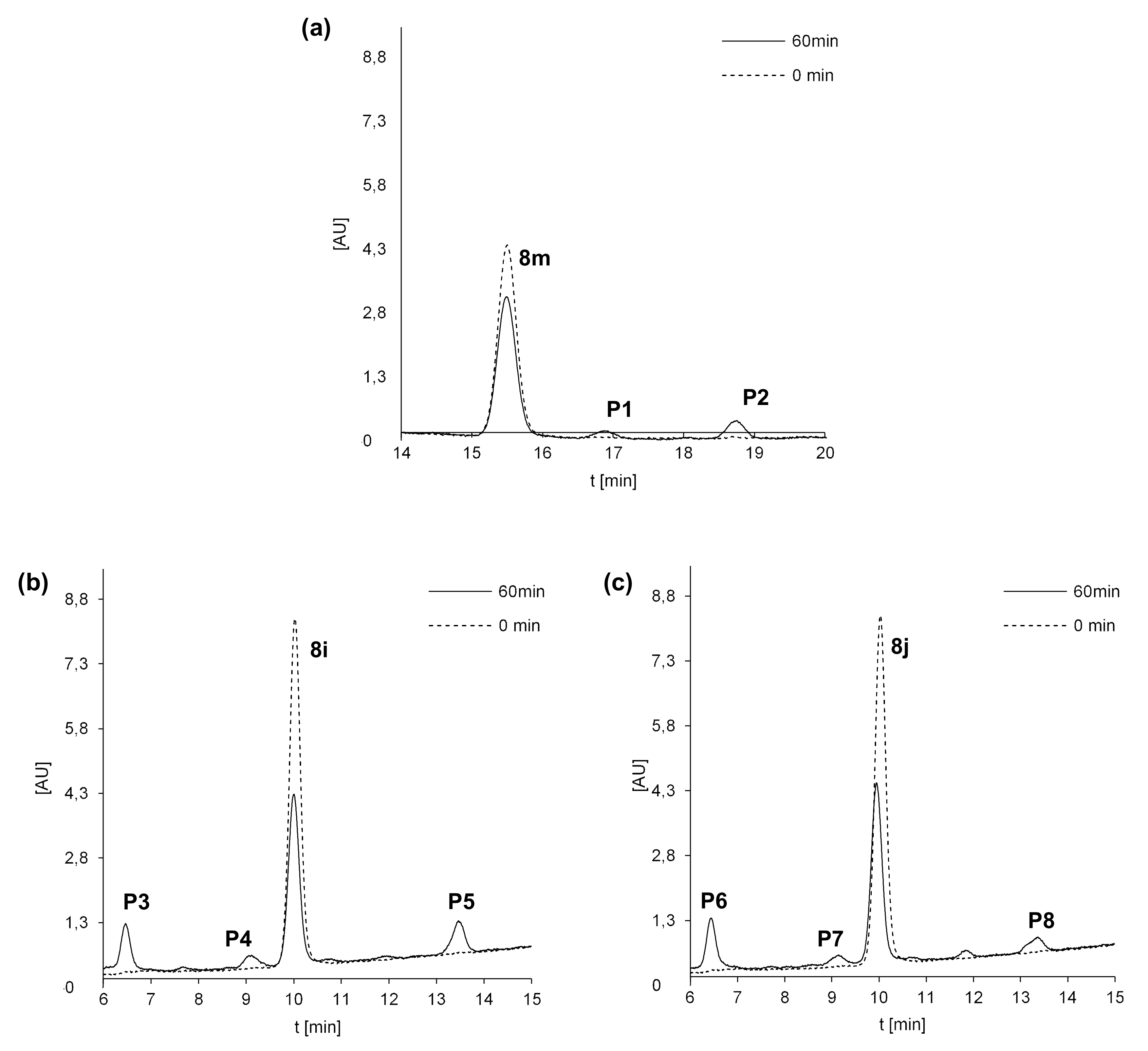
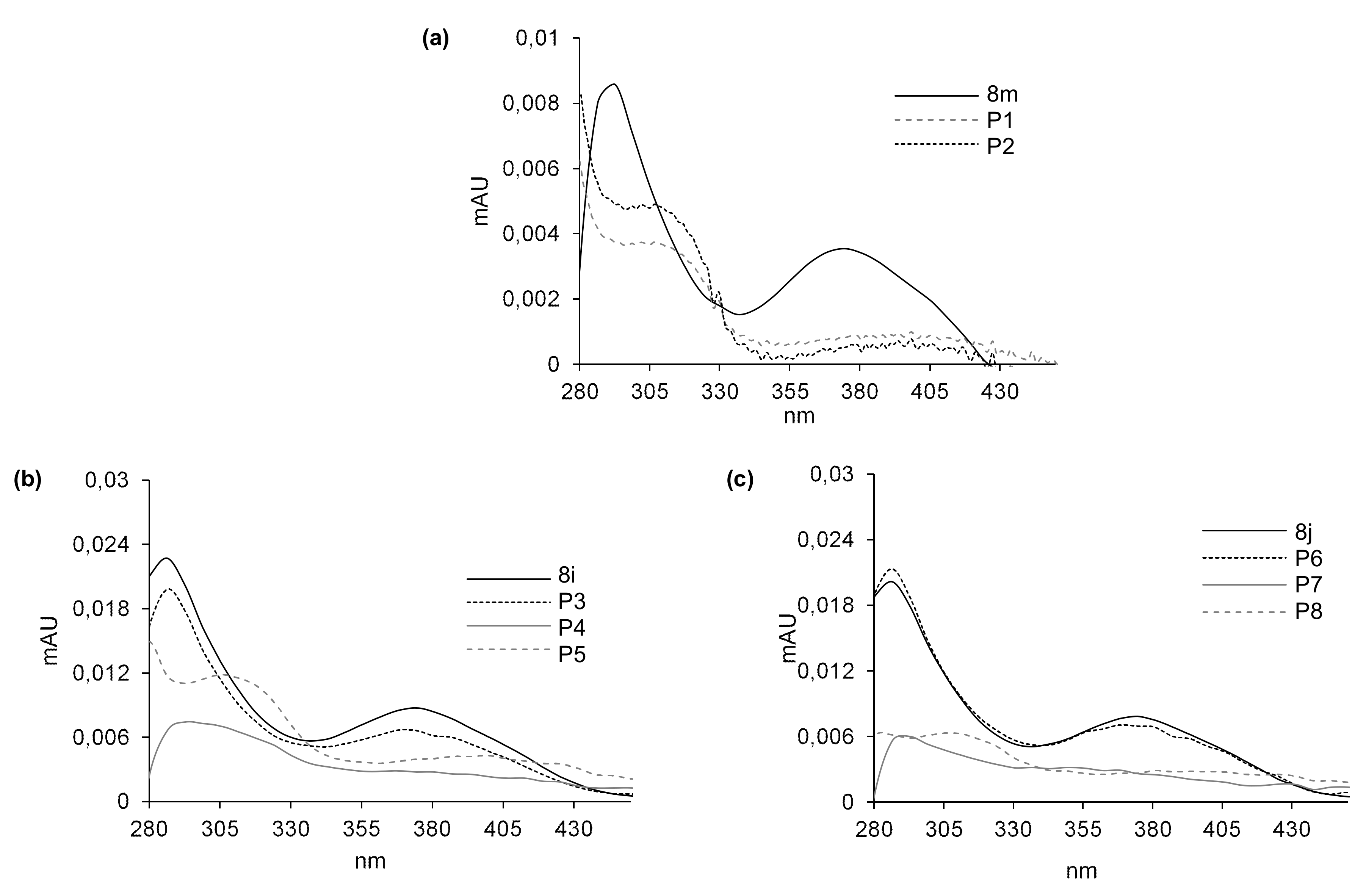
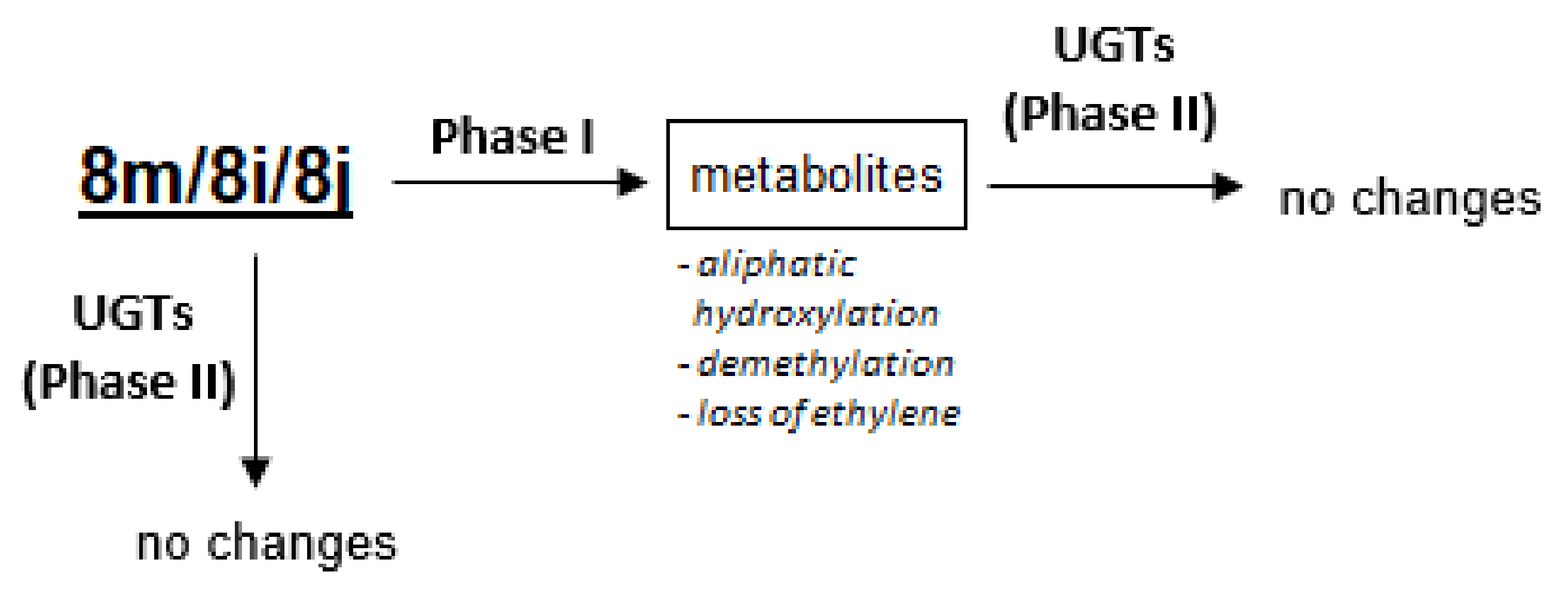
2.5. Susceptibility to Metabolic Transformation
2.5.1. Phase I Metabolism of 8m, 8i and 8j with Rat Liver Microsomes
2.5.2. Phase II Metabolism with Rat Liver Microsomes
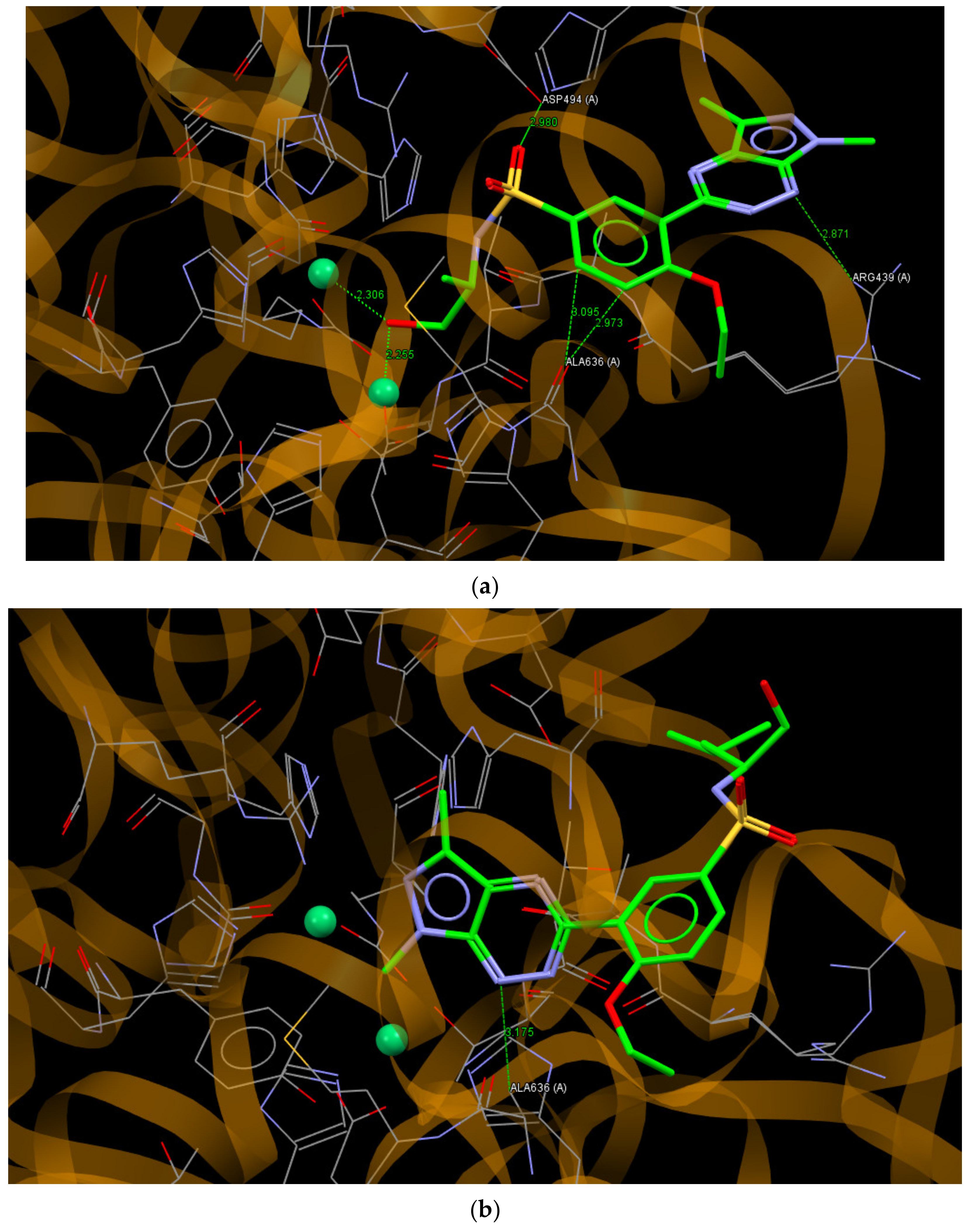
2.6. Molecular Docking
3. Conclusions
4. Experimental
4.1. Chemistry
4.1.1. Materials and Methods
4.1.2. Synthesis of Sulfonamides 8a–m
4.2. Lipophilicity and Protein Affinity Determination
4.2.1. UV-Vis Spectroscopy
4.2.2. RP-18 Chromatography
4.2.3. IAM Chromatography
4.2.4. Affinity Chromatography
4.2.5. Calibration of the Protein Columns
4.2.6. Computational Methods
4.3. Metabolic Transformation
4.3.1. Chemicals
4.3.2. Enzymes
4.3.3. Methods
Metabolism with Phase I Enzymes in Rat Liver Microsomes
Metabolism with Phase II, UGT and Enzymes in Rat Liver Microsomes
Metabolism with Phase I (NADPH) and Phase II (UDPGA) Enzymes in Rat Liver Microsomes
4.3.4. HPLC UV-Vis Analysis
4.3.5. Liquid Chromatography–Tandem Mass Spectrometry Analysis
4.4. Theoretical Calculations
4.5. Molecular Docking
4.6. Cell Cultures
4.7. MTT Assay
4.8. Kinase Inhibition Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindner, H.J.; Schaden, G. [Pyrazolo 4.3-e] as triazine, a new heterocyclic system from Pseudomonas fluorescens Var. Pseudoiodinum. Chem Ber. 1972, 105, 1949. [Google Scholar] [CrossRef]
- Hirata, K.; Nakagami, H.; Takashina, J.; Mahmud, T.; Kobayashi, M.; Ishida, T.; Miyamoto, K. Novel violet pigment, Nostocine A, an extracelluar metabolite from cyanobacterium Nostoc spongiaeforme. Heterocycles 1996, 43, 1513. [Google Scholar]
- Smirnov, V.V.; Kiprianova, E.A.; Garagulya, A.D.; Esipov, S.E.; Dovjenko, S.A. Fluviols, bicyclic nitrogen- rich antibiotics produced by Pseudomonas fluorescens. FEMS Microbiol. Lett. 1997, 153, 357. [Google Scholar] [CrossRef]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 5, 535. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925. [Google Scholar] [CrossRef]
- Kleeman, A.; Engel, J.; Kutscher, B.; Reichert, D. (Eds.) Pharmaceutical Substances, Syntheses, Patents, Applications; Thieme: Stuttgart, Germany; New York, NY, USA, 1999. [Google Scholar]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (VIAGRATM), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Disc. 2011, 10, 767. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Disc. 2008, 7, 168. [Google Scholar] [CrossRef]
- Khan, K.M.; Maharvi, G.M.; Perveen, S.; Khan, M.T.; Abdel-Jalil, R.J.; Shah, S.T.; Fecker, M.; Choudhary, M.I.; Voelter, W. Synthesis of methyl ether analogues of sildenafil (Viagra®) possessing tyrosinase inhibitory potential. Chem. Biodivers. 2005, 2, 470–476. [Google Scholar] [CrossRef]
- Khan, K.M.; Maharvi, G.M.; Khan, M.T.; Perveen, S.; Choudhary, M.I. A facile and improved synthesis of sildenafil (Viagra®) analogs through solid support microwave irradiation possessing tyrosinase inhibitory potential, their conformational analysis and molecular dynamics simulation studies. Mol. Divers. 2005, 9, 15–26. [Google Scholar] [CrossRef]
- Byth, K.F.; Culshaw, J.D.; Green, S.; Oakes, S.E.; Thomas, A.P. Imidazolo[1,2-a]pyridines. Part 2: SAR and optimization of a potent and selective class of cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 2249. [Google Scholar] [CrossRef] [PubMed]
- Shoaib Ahmad Shah, S.; Rivera, G.; Ashfaq, M. Recent advances in medicinal chemistry of sulfonamides. Rational design as anti-tumoral, anti-bacterial and anti-inflammatory agents. Mini Rev. Med. Chem. 2013, 13, 70–86. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Catalytic and inhibition mechanisms, distribution and physiological roles. In Carbonic Anhydrase: Its Inhibitors and Activators; CRC Press: Boca Raton, FL, USA, 2004; pp. 1–23. [Google Scholar]
- Supuran, C.T.; Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 2007, 15, 4336–4350. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Scozzafava, A.; Montero, J.L.; Supuran, C.T. Inhibition of carbonic anhydrase IX: A new strategy against cancer. Anticancer Agents Med. Chem. 2009, 9, 693–702. [Google Scholar] [CrossRef]
- Winum, S.J.Y.; Rami, M.; Scozzafava, A.; Montero, J.L.; Supuran, C.T. Carbonic Anhydrase IX: A new druggable target for the design of antitumor agents. Med. Res. Rev. 2008, 28, 445–463. [Google Scholar] [CrossRef] [PubMed]
- Pastoreková, S.; Pastorek, J. Cancer-related carbonic anhydrase isozymes and their inhibition. In Carbonic Anhydrase. Its Inhibitors and Activators; Supuran, C.T., Scozzafava, A., Conway, J., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 255–281. [Google Scholar]
- Mojzych, M.; Tarasiuk, P.; Kotwica-Mojzych, K.; Rafiq, M.; Seo, S.Y.; Nicewicz, M.; Fornal, E. Synthesis of chiral pyrazolo [4, 3-e][1, 2, 4] triazine sulfonamides with tyrosinase and urease inhibitory activity. J. Enzyme Inhib. Med. Chem. 2017, 32, 99–105. [Google Scholar] [CrossRef]
- Kaliszan, R.; Haber, P.; Baczek, T.; Siluk, D.; Valko, K. Lipophilicity and pKa estimates from gradient high-performance liquid chromatography. J. Chromatogr. A 2002, 965, 117–127. [Google Scholar] [CrossRef]
- Hollosy, F.; Valko, K.; Hersey, A.; Nunhuck, S.; Keri, G.; Bevan, C. Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. J. Med. Chem. 2006, 49, 6958–6971. [Google Scholar] [CrossRef] [PubMed]
- Rykowski, A.; Makosza, M. Reaction of 1, 2, 4-triazines with nitronate anions, direct nucleophilic acylation of 1, 2, 4-triazines. Tetrahedron Lett. 1984, 25, 4795. [Google Scholar] [CrossRef]
- Rykowski, A.; Lipinska, T. A Concise Route to a Key Intermediate in the Total Synthesis of Sempervirine1. Synth. Commun. 1996, 26, 4409. [Google Scholar] [CrossRef]
- Rykowski, A.; Mojzych, M.; Karczmarzyk, Z. A new synthesis of pyrazolo [4, 3-e][1, 2, 4] triazines via acid promoted ring closure of the phenylhydrazones of 5-acyl-1, 2, 4-triazines. Heterocycles 2000, 53, 2175. [Google Scholar] [CrossRef]
- Mojzych, M.; Rykowski, A. Transformations of phenylhydrazones of 5-acyl-1, 2, 4-triazines to pyrazolo [4, 3-e][1, 2, 4] triazines or 4-cyanopyrazole. J. Heterocycl. Chem. 2007, 44, 1003. [Google Scholar] [CrossRef]
- Alphonse, F.-A.; Suzenet, F.; Keromnes, A.; Lebret, B.; Guillaumet, G. Palladium-catalyzed 3-thiomethyltriazine-boronic acid cross coupling: Easy access to 3-substituted-1, 2, 4-triazines. Synlett 2002, 3, 447. [Google Scholar] [CrossRef]
- Mojzych, M.; Kubacka, M.; Mogilski, S.; Filipek, B.; Fornal, E. Relaxant effects of selected sildenafil analogues in the rat aorta. J. Enzyme Inhib. Med. Chem. 2016, 31, 381. [Google Scholar]
- Mojzych, M.; Karczmarzyk, Z.; Wysocki, W.; Ceruso, M.; Supuran, C.T.; Krystof, V.; Urbańczyk-Lipkowska, Z.; Kalicki, P. New approaches to the synthesis of sildenafil analogues and their enzyme inhibitory activity. Bioorg. Med. Chem. 2015, 23, 1421. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Ceruso, M.; Bielawska, A.; Bielawski, K.; Fornal, E.; Supuran, C.T. New pyrazolo [4, 3-e][1, 2, 4] triazine sulfonamides as carbonic anhydrase inhibitors. Bioorg. Med. Chem. 2015, 23, 3674. [Google Scholar] [CrossRef] [PubMed]
- Mojzych, M.; Bielawska, A.; Bielawski, K.; Ceruso, M.; Supuran, C.T. Pyrazolo [4, 3-e][1, 2, 4] triazine sulfonamides as carbonic anhydrase inhibitors with antitumor activity. Bioorg. Med. Chem. 2014, 22, 2643. [Google Scholar] [CrossRef]
- Soczewinski, E.; Wachtmeister, C.A. The relation between the composition of certain ternary two-phase solvent systems and RM values. J. Chromatogr. A 1962, 7, 311. [Google Scholar] [CrossRef]
- Janicka, M.; Kwietniewski, L.; Matysiak, J. A new method for estimating log kw values and solute biological activity. JPC-J. Planar Chromat. 2000, 13, 285. [Google Scholar]
- Niewiadomy, A.; Zabinska, A.; Matysiak, J.; Rozylo, J.K. Influence of modifier and molecular structure of some dihydroxythiobenzanilides on retention in reversed-phase high-performance thin-layer chromatography. J. Chromatogr. A 1997, 791, 237. [Google Scholar] [CrossRef]
- Valko, K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299. [Google Scholar] [CrossRef]
- Ong, S.; Pidgeon, C. Thermodynamics of solute partitioning into immobilized artificial membranes. Anal. Chem. 1995, 67, 2119. [Google Scholar]
- Mojzych, M.; Bernat, Z.; Karczmarzyk, Z.; Matysiak, J.; Fruziński, A. Synthesis, Structural Characterization, and Biological Activity of New Pyrazolo [4, 3-e][1, 2, 4] triazine Acyclonucleosides. Molecules 2020, 25, 221. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comp. Sci. 1987, 27, 21. [Google Scholar] [CrossRef]
- Petrauskas, A.A.; Kolovanov, E.A. ACD/Log P method description. Perspect. Drug Discov. Des. 2000, 19, 99. [Google Scholar] [CrossRef]
- Janicka, M.; Sztanke, M.; Sztanke, K. Reversed-phase liquid chromatography with octadecylsilyl, immobilized artificial membrane and cholesterol columns in correlation studies with in silico biological descriptors of newly synthesized antiproliferative and analgesic active compounds. J. Chromatogr. A 2013, 1318, 91. [Google Scholar] [CrossRef]
- Pyka, A.; Buska, M.; Zachariasz, M. A comparison of theoretical methods of calculation of partition coefficients for selected drugs. Acta Pol. Pharm. 2006, 63, 159. [Google Scholar]
- Barbato, F.; Cirocco, V.; Grumetto, L.; la Rotonda, M.I. Comparison between immobilized artificial membrane (IAM) HPLC data and lipophilicity in n-octanol for quinolone antibacterial agents. Eur. J. Pharm. Sci. 2007, 31, 288–297. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reyncilds, D.P. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J. Pharm. Sci. 2003, 92, 2236. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.M.; Ancellin, N.; Beaufils, B.; Bingham, R.P.; Borthwick, J.A.; Boullay, A.B.; Boursier, E.; Carter, P.S.; Chung, C.W.; Churcher, I.; et al. The discovery of in vivo active mitochondrial branched-chain aminotransferase (BCATm) inhibitors by hybridizing fragment and HTS hits. J. Med. Chem. 2015, 58, 7140. [Google Scholar] [CrossRef] [PubMed]
- Noctor, T.A.; Diaz-Perez, M.J.; Wainer, I.W. Use of a human serum albumin-based stationary phase for high-performance liquid chromatography as a tool for the rapid determination of drug-plasma protein binding. J. Pharm. Sci. 2015, 82, 675. [Google Scholar] [CrossRef]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; Choudhary, M.I.; Betzel, C. The First Jack Bean Urease (Canavalia Ensiformis) Complex Obtained at 1.52 Resolution. 2012. Available online: https://dx.doi.org/10.2210/pdb4h9m/pdb (accessed on 20 March 2009).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Pople, Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- HyperChem. Release 8.0.10 for Windows Molecular Modeling System; Hypercube Inc.: Gainesville, FL, USA, 2011. [Google Scholar]
- Frisch, A.; Dennington, R.D., II; Keith, T.A.; Millam, J.; Nielsen, A.B.; Holder, A.J.; Hiscocks, J. Gauss View Reference; Version 4; Gaussian Inc.: Wallingford, CT, USA, 2007. [Google Scholar]
- Jones, G.G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein−Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Carmichael, J.; Degraff, W.; Gazdar, A.; Minna, J.; Mitchell, J. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936. [Google Scholar] [PubMed]
- Mojzych, M.; Subertová, V.; Bielawska, A.; Bielawski, K.; Bazgier, V.; Berka, K.; Gucký, T.; Fornal, E.; Krystof, V. Synthesis and kinase inhibitory activity of new sulfonamide derivatives of pyrazolo[4,3-e][1,2,4]triazines. Eur. J. Med. Chem. 2014, 78, 217. [Google Scholar] [CrossRef]
- Zatloukal, M.; Jorda, R.; Gucky, T.; Reznickova, E.; Voller, J.; Pospisil, T.; Malinkova, V.; Adamcova, H.; Krystof, V.; Strnad, M. Synthesis and in vitro biological evaluation of 2, 6, 9-trisubstituted purines targeting multiple cyclin-dependent kinases. Eur. J. Med. Chem. 2013, 61, 61–72. [Google Scholar] [CrossRef]
- Jorda, R.; Havlicek, L.; McNae, I.W.; Walkinshaw, M.D.; Voller, J.; Sturc, A.; Navratilová, J.; Kuzma, M.; Mistrik, M.; Bartek, J.; et al. Pyrazolo [4, 3-d] pyrimidine bioisostere of roscovitine: Evaluation of a novel selective inhibitor of cyclin-dependent kinases with antiproliferative activity. J. Med. Chem. 2011, 54, 2980–2993. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | φ1 | φ2 | φ3 | φ4 | φ5 |
|---|---|---|---|---|---|
| 8a | 48.34 | −176.79 | 177.69 | −61.88 | −86.87 |
| 8b | 48.12 | −176.14 | 177.58 | −79.54 | −65.17 |
| 8c | 48.49 | −176.67 | 177.30 | −71.34 | −91.70 |
| 8d | 48.27 | −176.62 | 177.40 | −73.26 | −105.64 |
| 8e | 48.38 | −175.89 | 177.14 | −82.67 | −64.24 |
| 8f | 47.82 | −176.36 | 177.66 | −74.63 | −92.87 |
| 8g | 48.02 | −176.50 | 177.62 | −70.19 | −91.80 |
| 8h | 48.24 | −176.20 | 178.03 | −84.06 | −65.37 |
| 8i | 48.12 | −175.91 | 177.43 | −77.67 | −52.71 |
| 8j | 48.21 | −175.98 | 177.62 | −76.04 | −62.26 |
| 8k | 48.82 | −176.71 | 177.24 | −57.63 | −86.19 |
| 8l | 48.99 | −176.38 | 177.10 | −56.25 | −93.33 |
| 8m | −47.34 | −176.31 | 178.33 | −89.01 | 69.02 |
| Comp. | ET | MD | EHOMO | ELUMO | ΔE |
|---|---|---|---|---|---|
| 8a | −1,059,794.579 | 7.896 | −154.284 | −64.357 | 89.927 |
| 8b | −1,059,793.306 | 7.177 | −151.234 | −62.386 | 88.848 |
| 8c | −1,109,145.745 | 7.857 | −153.795 | −64.086 | 89.709 |
| 8d | −1,109,143.310 | 6.350 | −153.380 | −64.024 | 89.356 |
| 8e | −1,059,794.523 | 6.287 | −153.619 | −64.363 | 89.256 |
| 8f | −1,059,795.508 | 7.522 | −153.631 | −63.905 | 89.726 |
| 8g | −1,107,011.827 | 7.035 | −153.387 | −63.579 | 89.808 |
| 8h | −1,107,007.271 | 7.434 | −151.253 | −63.114 | 88.139 |
| 8i | −1,133,819.771 | 8.453 | −155.050 | −65.474 | 89.576 |
| 8j | −1,133,818.278 | 6.853 | −151.467 | −62.738 | 88.729 |
| 8k | −1,131,683.174 | 8.171 | −152.809 | −64.639 | 88.170 |
| 8l | −1,084,469.221 | 7.549 | −150.193 | −62.838 | 87.355 |
| 8m | −1,095,904.170 | 6.682 | −145.160 | −62.856 | 82.304 |
| Atom | 8a | 8b | 8c | 8d | 8e | 8f | 8g | 8h | 8i | 8j | 8k | 8l | 8m |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N1 | −0.202 | −0.208 | −0.202 | −0.202 | −0.202 | −0.202 | −0.202 | −0.202 | −0.201 | −0.208 | −0.201 | −0.202 | −0.203 |
| N2 | −0.232 | −0.229 | −0.232 | −0.231 | −0.231 | −0.231 | −0.232 | −0.231 | −0.231 | −0.229 | −0.231 | −0.231 | −0.231 |
| C3 | +0.378 | +0.378 | +0.378 | +0.378 | +0.378 | +0.378 | +0.377 | +0.380 | +0.378 | +0.378 | +0.380 | +0.381 | +0.379 |
| N4 | −0.412 | −0.410 | −0.412 | −0.413 | −0.413 | −0.416 | −0.416 | −0.412 | −0.414 | −0.412 | −0.414 | −0.413 | −0.413 |
| C5 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.113 | +0.112 | +0.112 | +0.113 |
| C6 | +0.318 | +0.322 | +0.318 | +0.318 | +0.318 | +0.318 | +0.318 | +0.317 | +0.318 | +0.322 | +0.317 | +0.317 | +0.317 |
| N7 | −0.226 | −0.231 | −0.226 | −0.226 | −0.226 | −0.226 | −0.226 | −0.226 | −0.225 | −0.232 | −0.226 | −0.225 | −0.225 |
| N8 | −0.235 | −0.235 | −0.235 | −0.235 | −0.234 | −0.235 | −0.235 | −0.235 | −0.234 | −0.235 | −0.235 | −0.236 | −0.235 |
| C9 | +0.157 | +0.159 | +0.157 | +0.157 | +0.157 | +0.158 | +0.158 | +0.156 | +0.157 | +0.159 | +0.156 | +0.156 | +0.157 |
| O18 | −0.537 | −0.539 | −0.537 | −0.536 | −0.537 | −0.535 | −0.536 | −0.538 | −0.538 | −0.539 | −0.538 | −0.540 | −0.530 |
| S21 | +2.207 | +2.227 | +2.212 | +2.217 | +2.217 | +2.209 | +2.209 | +2.220 | +2.225 | +2.230 | +2.224 | +2.219 | +2.238 |
| O22 | −0.904 | −0.913 | −0.907 | −0.911 | −0.913 | −0.907 | −0.961 | −0.913 | −0.913 | −0.914 | −0.908 | −0.915 | −0.917 |
| O23 | −0.952 | −0.925 | −0.950 | −0.950 | −0.921 | −0.952 | −0.959 | −0.927 | −0.940 | −0.927 | −0.962 | −0.931 | −0.920 |
| N24 | −0.875 | −0.890 | −0.885 | −0.893 | −0.865 | −0.874 | −0.886 | −0.875 | −0.896 | −0.895 | −0.916 | −0.913 | −0.750 |
| Compd. | MTT Assay, IC50 (µM) a | ||||
|---|---|---|---|---|---|
| CDK2/E | Abl | K562 | MCF-7 | p53 | |
| 8a | >100 | >100 | >100 | >100 | >100 |
| 8b | >100 | >100 | >100 | >100 | >100 |
| 8c | >100 | >100 | >100 | >100 | >100 |
| 8d | >100 | >100 | >100 | >100 | >100 |
| 8e | >100 | >100 | >100 | >100 | >100 |
| 8f | >100 | >100 | >100 | >100 | >100 |
| 8g | >100 | >100 | >100 | >100 | >100 |
| 8h | >100 | >100 | >100 | >100 | >100 |
| 8i | >100 | >100 | >100 | >100 | >100 |
| 8j | >100 | >100 | >100 | >100 | >100 |
| 8k | >100 | >100 | >100 | >100 | >100 |
| 8l | >100 | >100 | >100 | >100 | >100 |
| chlorambucil | n.a. | n.a. | 84 ± 6 | 97 ± 2 | |
| imatinib | >100 | 0.3 | 13 ± 2 | n.a. | |
| roscovitine | 0.1 | >100 | 42 | 11 | |
| No. | RP-18 HPLC, pH = 7.4 | IAM HPLC, pH = 7.4 | Δ log kw | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | log kw | r2 | n | φ | S | log kw | r2 | n | ||
| 8a | 4.8991 | 2.5078 | 0.9967 | 13 | 30–90 | 6.0158 | 1.3981 | 0.9879 | 10 | 1.1097 |
| 8b | 4.8457 | 2.4918 | 0.9977 | 13 | 30–90 | 5.8731 | 1.3369 | 0.9929 | 10 | 1.1549 |
| 8c | 5.1604 | 2.9189 | 0.998 | 11 | 40–90 | 5.7249 | 1.6166 | 0.9924 | 8 | 1.3023 |
| 8d | 4.9656 | 2.8108 | 0.9957 | 10 | 40–85 | 5.7839 | 1.6304 | 0.9913 | 8 | 1.1804 |
| 8e | 4.7800 | 2.5211 | 0.9862 | 13 | 30–90 | 5.5150 | 1.3230 | 0.9951 | 10 | 1.1981 |
| 8f | 4.8777 | 2.5295 | 0.9963 | 13 | 30–90 | 5.9269 | 1.3995 | 0.9870 | 10 | 1.1300 |
| 8g | 4.6290 | 2.1677 | 0.9905 | 13 | 30–90 | 5.1630 | 1.0874 | 0.9867 | 9 | 1.0803 |
| 8h | 4.7517 | 2.2397 | 0.9892 | 12 | 30–85 | 5.3347 | 1.1156 | 0.9903 | 10 | 1.1241 |
| 8i | 5.6107 | 3.3825 | 0.9963 | 11 | 40–90 | 6.6527 | 2.0414 | 0.9829 | 9 | 1.3411 |
| 8j | 5.6704 | 3.2371 | 0.9960 | 10 | 40–85 | 6.1622 | 1.9301 | 0.9925 | 8 | 1.3070 |
| 8k | 4.8948 | 2.4162 | 0.9954 | 12 | 30–85 | 5.4320 | 1.2184 | 0.9908 | 10 | 1.1978 |
| 8l | 5.0611 | 2.7706 | 0.994 | 11 | 35–85 | 5.7380 | 1.5155 | 0.9901 | 9 | 1.2551 |
| 8m | 3.7686 | 2.5756 | 0.9984 | 7 | 30–85 | 5.1248 | 1.5911 | 0.9816 | 6 | 0.9845 |
| No. | HBDH | M log P | Clog P | ACD log P | tPSA | CMR | pK a |
|---|---|---|---|---|---|---|---|
| 8a | 2 | 0.435 | 0.299 | 1.46 | 132.12 | 10.27 | 1.01 |
| 8b | 2 | 0.435 | 0.299 | 1.46 | 132.12 | 10.27 | 1.01 |
| 8c | 2 | 0.905 | 1.227 | 2.34 | 132.12 | 11.19 | 1.01 |
| 8d | 2 | 0.905 | 1.227 | 2.34 | 132.12 | 11.19 | 1.01 |
| 8e | 2 | 0.435 | 0.299 | 1.46 | 132.12 | 10.27 | 1.01 |
| 8f | 2 | 0.435 | 0.299 | 1.46 | 132.12 | 10.27 | 1.01 |
| 8g | 3 | −0.289 | −0.157 | 0.50 | 152.35 | 10.42 | 1.01 |
| 8h | 3 | −0.289 | −0.157 | 0.50 | 152.35 | 10.42 | 1.01 |
| 8i | 2 | 1.132 | 1.755 | 2.87 | 132.12 | 11.66 | 1.01 |
| 8j | 2 | 1.132 | 1.755 | 2.87 | 132.12 | 11.66 | 1.01 |
| 8k | 3 | −0.051 | 0.248 | 1.28 | 152.35 | 10.88 | 1.01 |
| 8l | 2 | 0.672 | 0.698 | 1.81 | 132.12 | 10.73 | 1.01 |
| 8m | 0 | 0.905 | 1.122 | 2.27 | 102.54 | 11.23 | 1.01/5.96 |
| HSA | AGP | |||||
|---|---|---|---|---|---|---|
| log kHSA | log KHSA | % HSA | log kAGP | log KAGP | % AGP | |
| 8a | −0.78.4 | −0.420 | 27.8 | −1.841 | −1.647 | 2.3 |
| 8b | −0.822 | −0.459 | 26,1 | −0.6199 | −0.396 | 28.9 |
| 8c | −0.442 | −0.075 | 46.1 | −0.519 | −0.293 | 34.1 |
| 8d | −0.782 | −0.418 | 27.9 | −0.483 | −0.257 | 36.0 |
| 8e | −2.052 | −1.702 | 2.0 | −0.966 | −0.751 | 15.2 |
| 8f | −1.771 | −1.418 | 3.7 | −0.986 | −0.771 | 14.6 |
| 8g | −0.926 | −0.564 | 21.6 | −0.762 | −0.543 | 22.5 |
| 8h | −1.248 | −0.890 | 11.5 | −1.675 | −1.477 | 3.3 |
| 8i | −0.205 | 0.165 | 60.0 | −0.297 | −0.066 | 46.6 |
| 8j | −0.186 | 0.1831 | 61.0 | −0.384 | −0.155 | 41.6 |
| 8k | −0.917 | −0.555 | 22.0 | −1.646 | −1.447 | 3.5 |
| 8l | −0.694 | −0.329 | 32.2 | −0.494 | −0.268 | 35.4 |
| 8m | −0.197 | 0.173 | 60.4 | −0.222 | 0.010 | 51.0 |
| Compd. | ESI-MS m/z | |
|---|---|---|
| 8m | 432.1 | [8m + H+] |
| P1 | 448.1 | [8m + 16 + H+] |
| P2 | 404.1 | [8m-28 + H+] |
| 8i | 449.1 | [8i + H+] |
| P3 | 465.1 | [8i + 16 + H+] |
| P4 | 435.1 | [8i-14 + H+] |
| P5 | 421.1 | [8i-28 + H+] |
| 8j | 449.1 | [8j + H+] |
| P6 | 465.1 | [8j + 16 + H+] |
| P7 | 435.1 | [8j-14 + H+] |
| P8 | 421.1 | [8j-28 + H+] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernat, Z.; Mieszkowska, A.; Mazerska, Z.; Matysiak, J.; Karczmarzyk, Z.; Kotwica-Mojzych, K.; Mojzych, M. Chiral Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides—Their Biological Activity, Lipophilicity, Protein Affinity, and Metabolic Transformations. Appl. Sci. 2021, 11, 2660. https://doi.org/10.3390/app11062660
Bernat Z, Mieszkowska A, Mazerska Z, Matysiak J, Karczmarzyk Z, Kotwica-Mojzych K, Mojzych M. Chiral Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides—Their Biological Activity, Lipophilicity, Protein Affinity, and Metabolic Transformations. Applied Sciences. 2021; 11(6):2660. https://doi.org/10.3390/app11062660
Chicago/Turabian StyleBernat, Zofia, Anna Mieszkowska, Zofia Mazerska, Joanna Matysiak, Zbigniew Karczmarzyk, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2021. "Chiral Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides—Their Biological Activity, Lipophilicity, Protein Affinity, and Metabolic Transformations" Applied Sciences 11, no. 6: 2660. https://doi.org/10.3390/app11062660
APA StyleBernat, Z., Mieszkowska, A., Mazerska, Z., Matysiak, J., Karczmarzyk, Z., Kotwica-Mojzych, K., & Mojzych, M. (2021). Chiral Pyrazolo[4,3-e][1,2,4]triazine Sulfonamides—Their Biological Activity, Lipophilicity, Protein Affinity, and Metabolic Transformations. Applied Sciences, 11(6), 2660. https://doi.org/10.3390/app11062660