Theoretical Prediction of P-Triphenylene-Graphdiyne as an Excellent Anode Material for Li, Na, K, Mg, and Ca Batteries




Abstract
1. Introduction
2. Computational Method
3. Results and Discussion
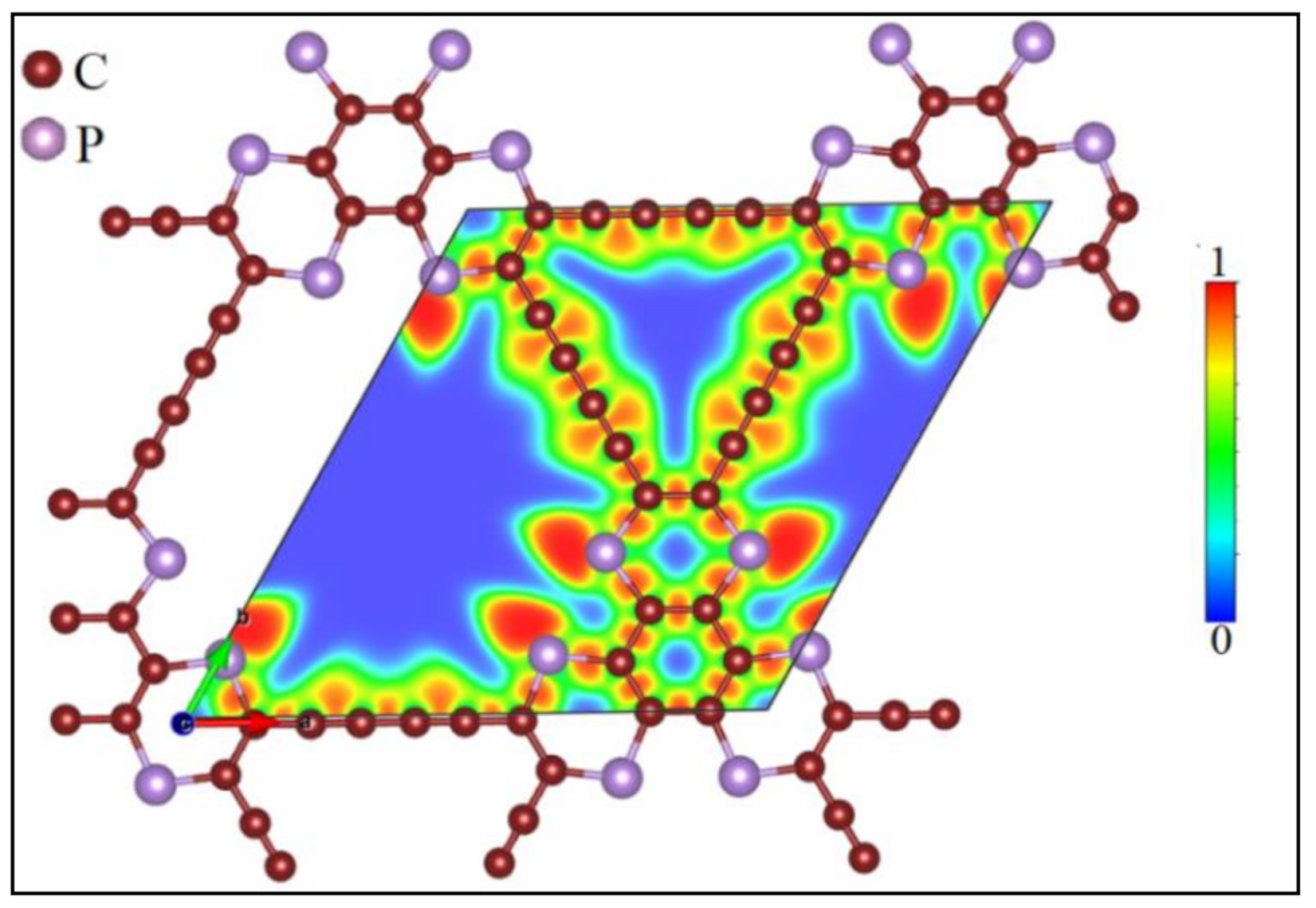
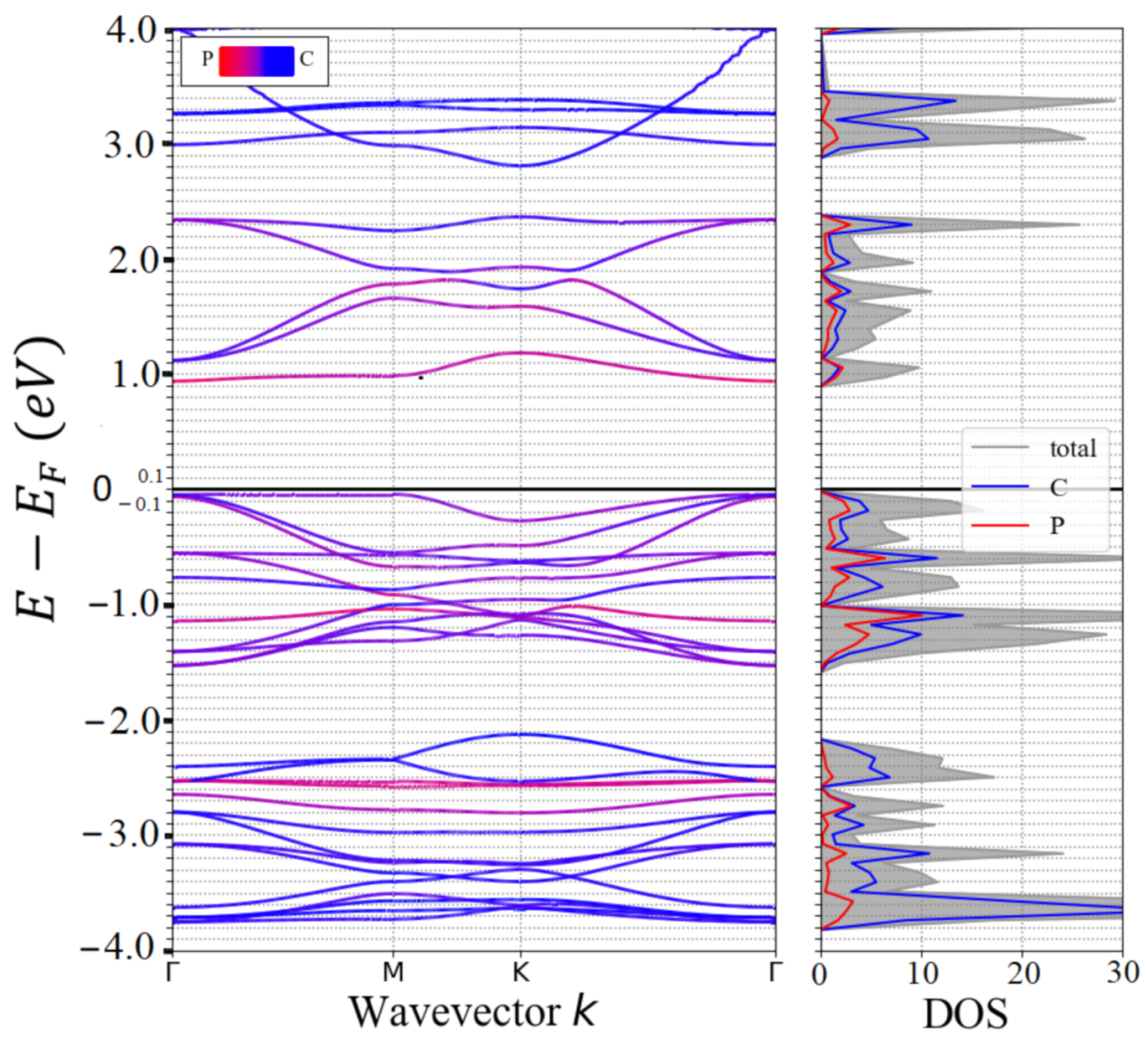
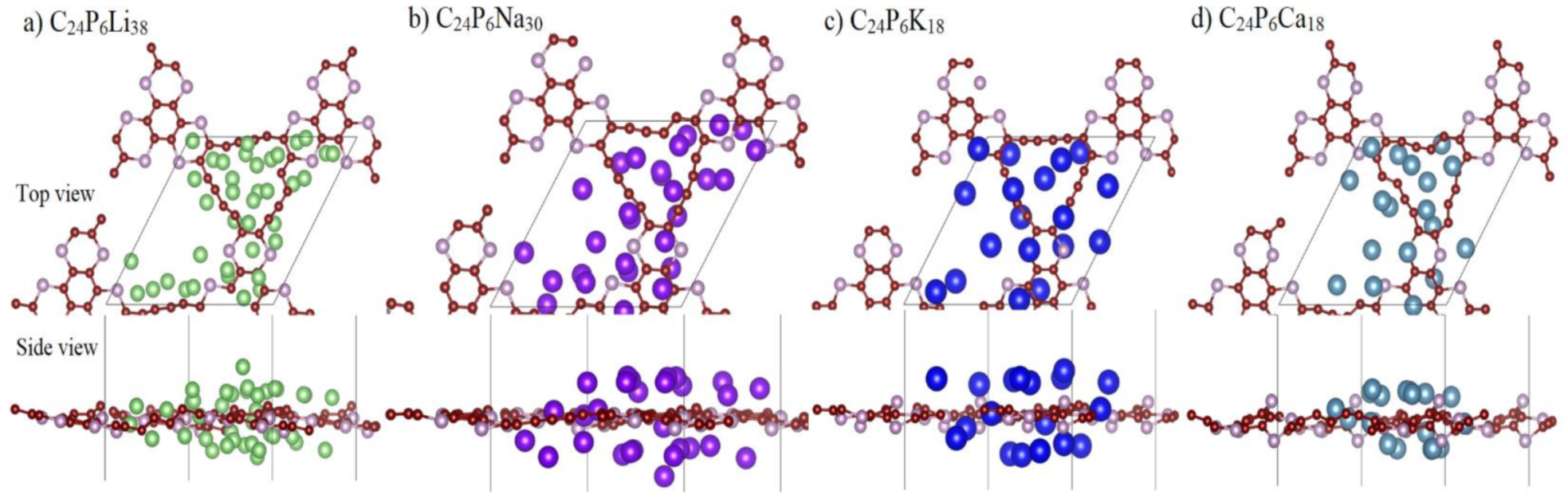
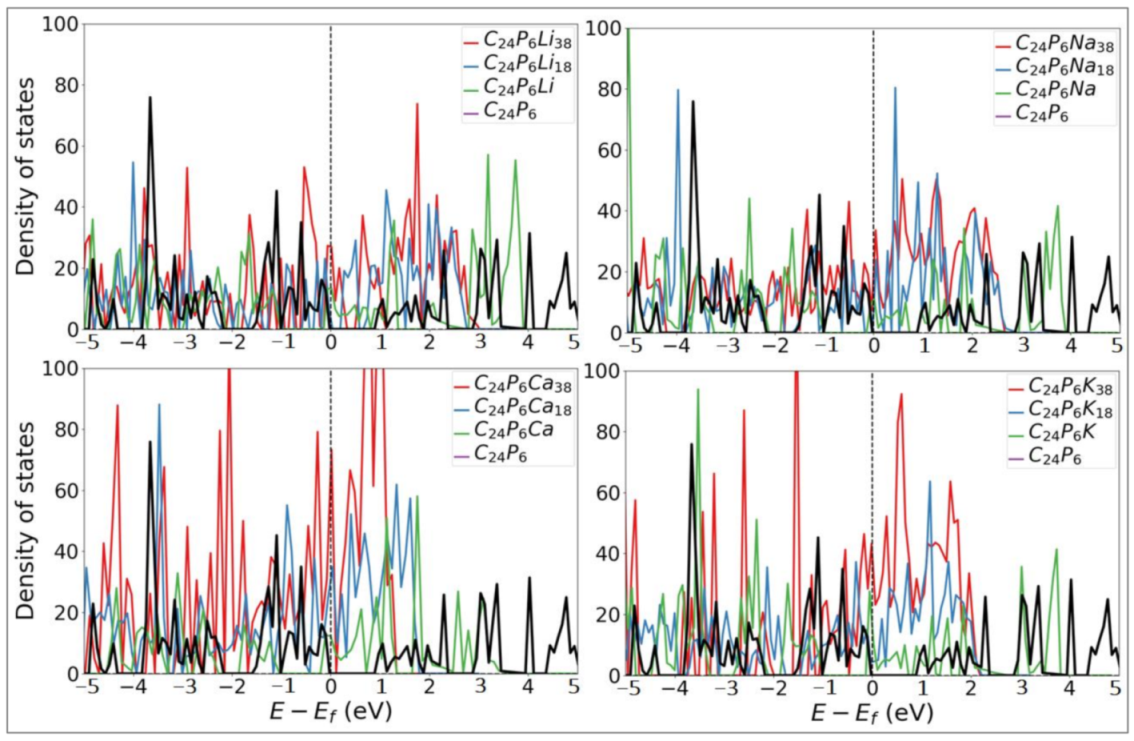
3.1. Lattice Structure and Electronic Properties
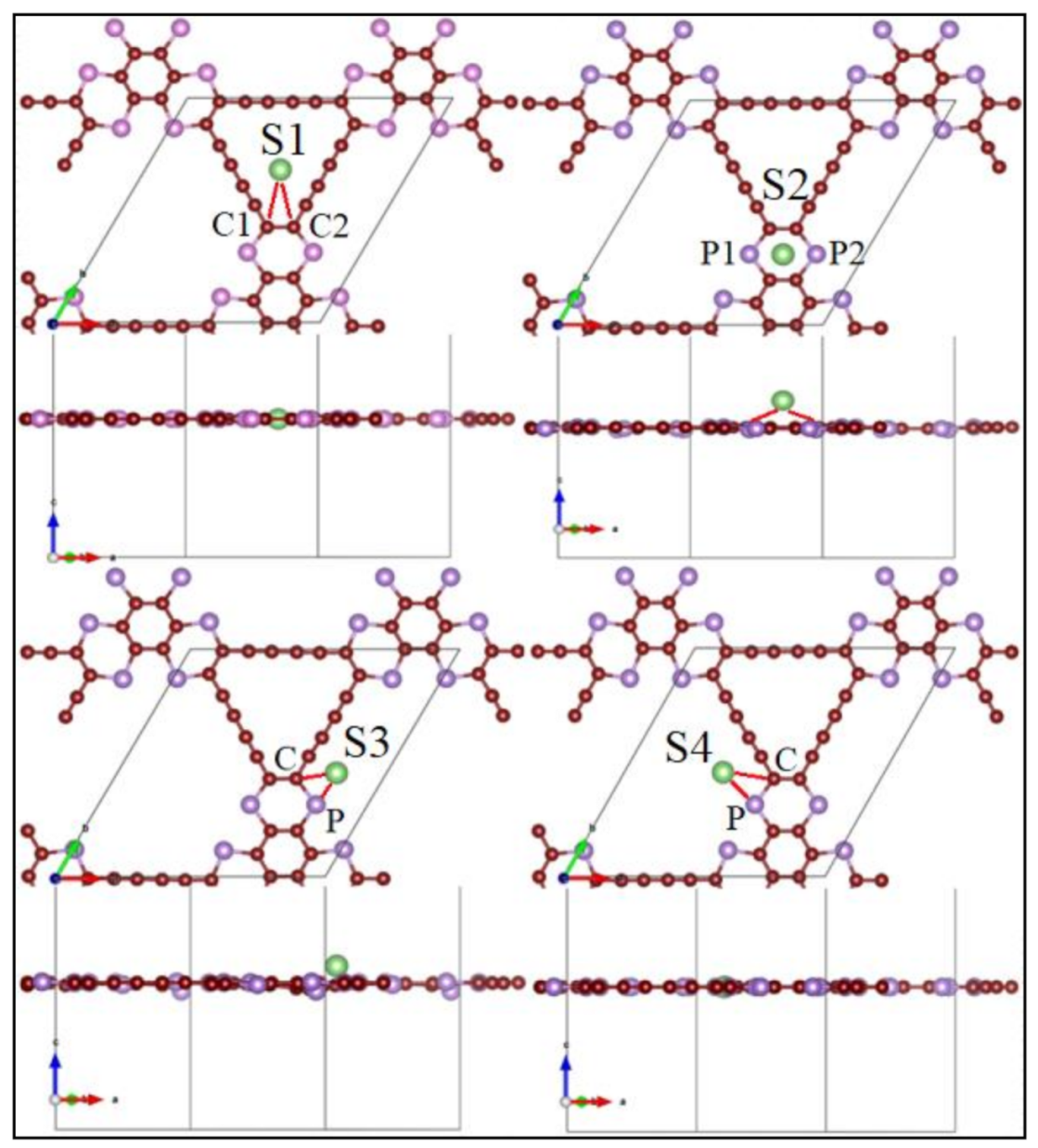
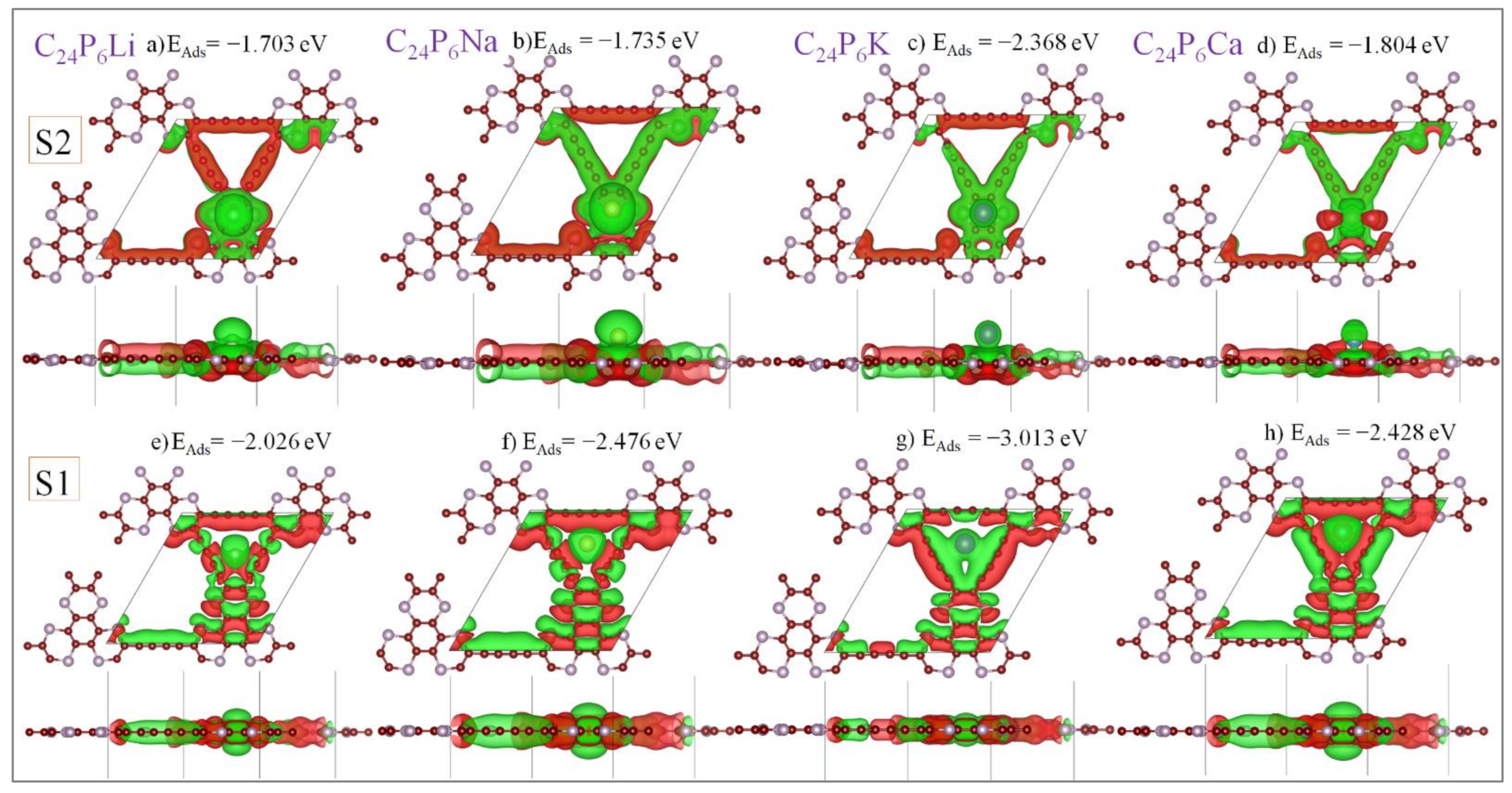
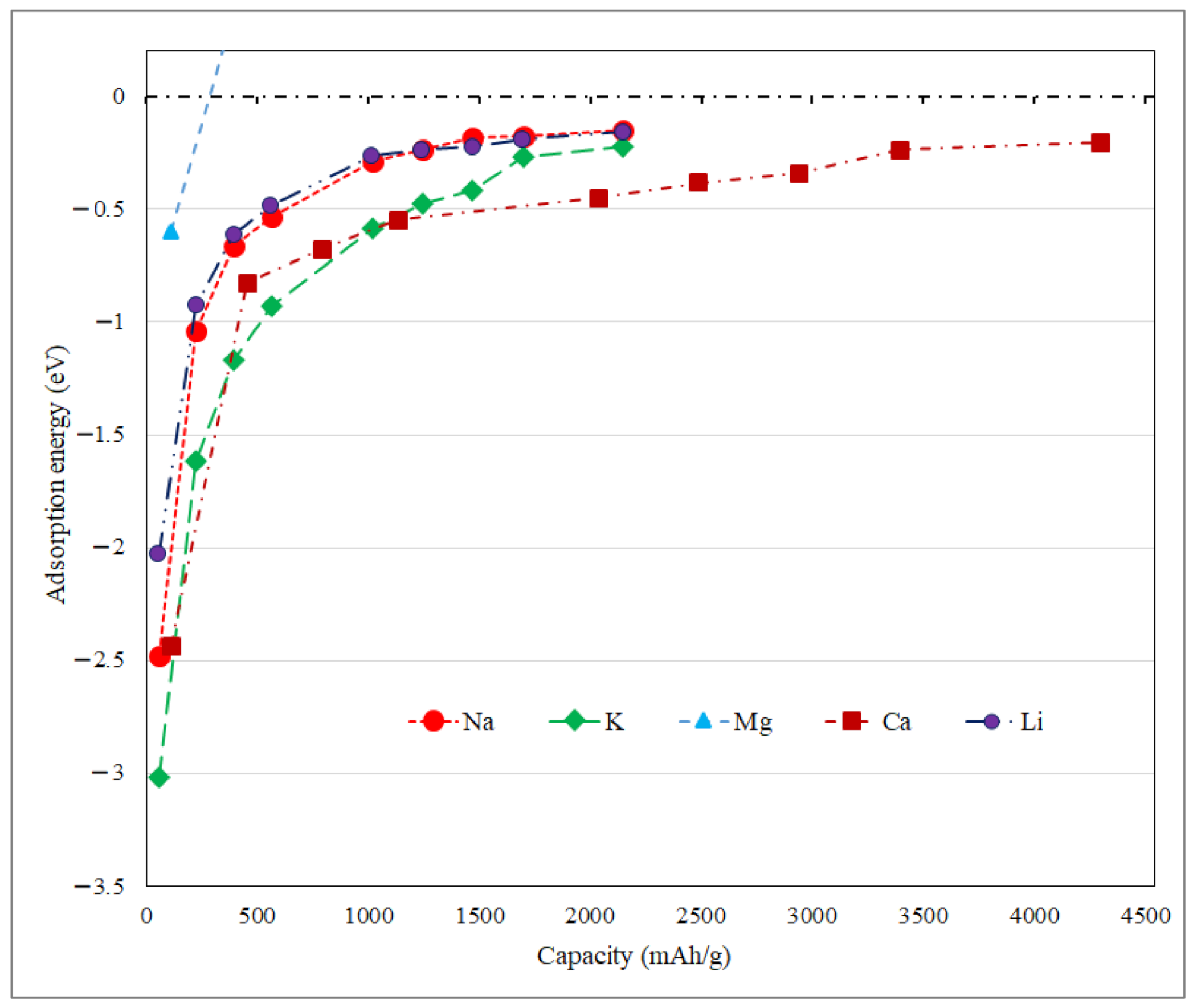
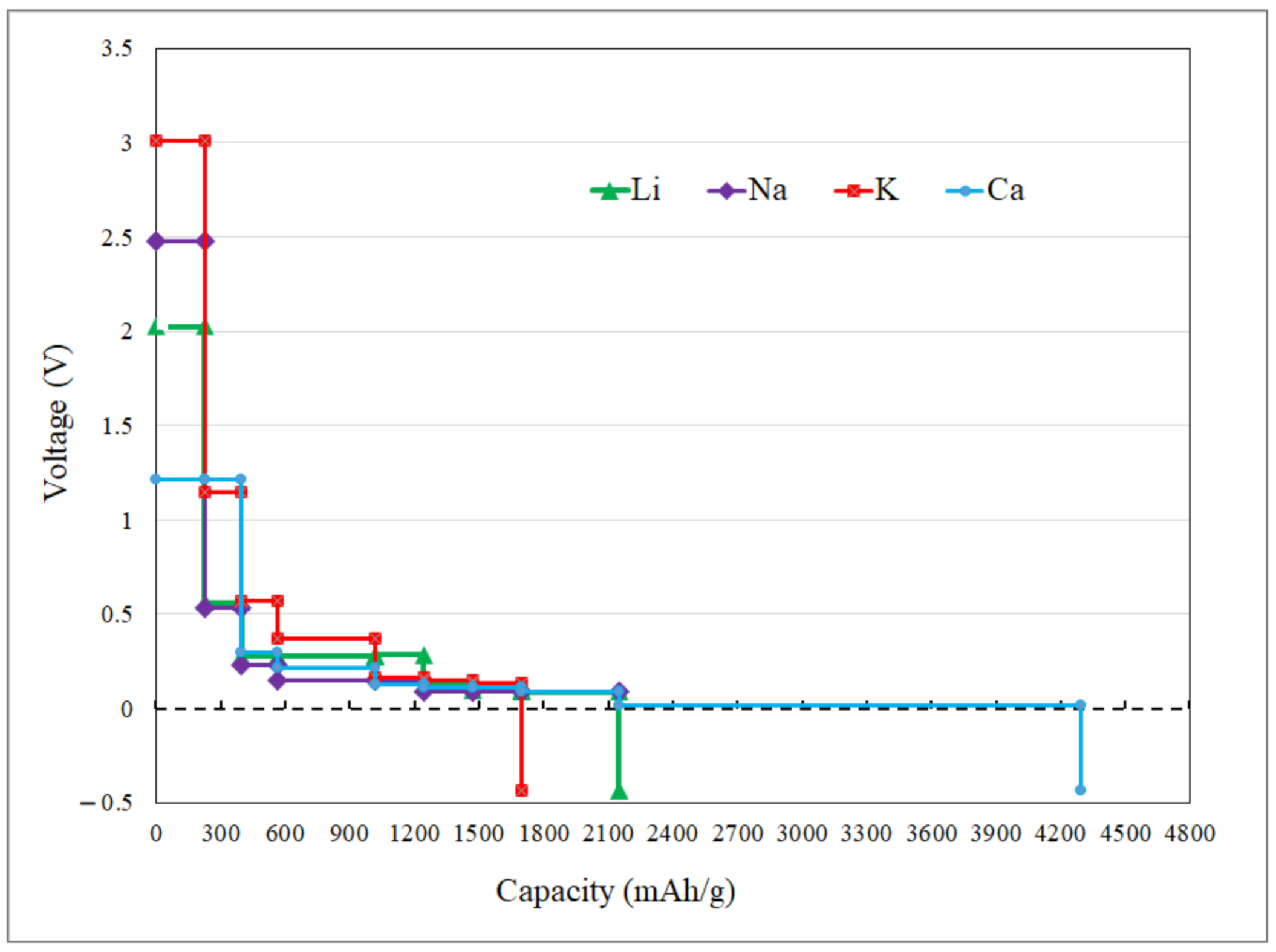
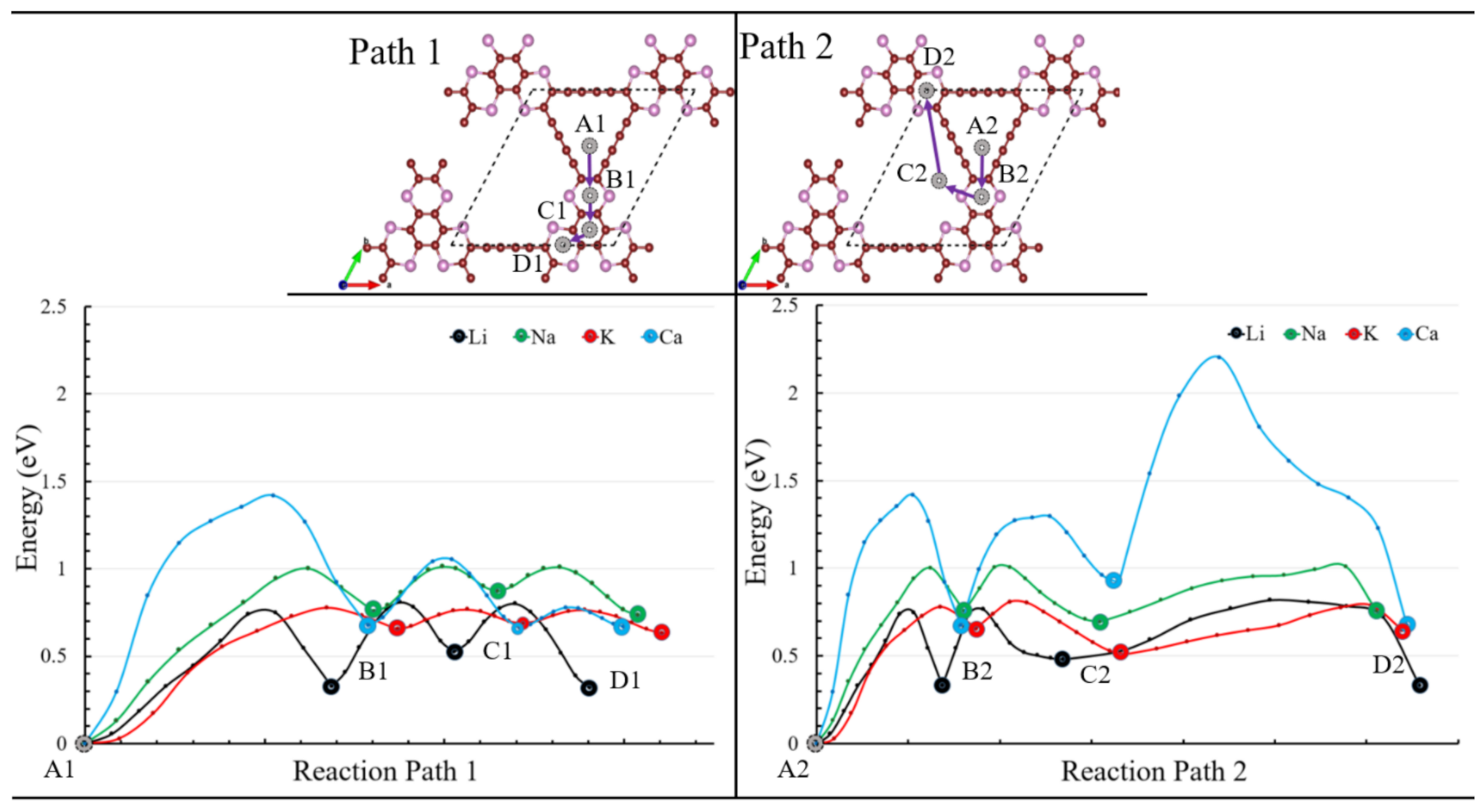
3.2. Application as Anode Materials for Li, Na, K, Mg and Ca-Ions Batteries
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, W.; Zhang, J.; Li, H. Batteries with high theoretical energy densities. Energy Storage Mater. 2020, 26, 46–55. [Google Scholar] [CrossRef]
- Zhu, K.; Li, Q.; Xue, Z.; Yu, Q.; Liu, X.; Shan, Z.; Liu, K. Mesoporous TiO2 Spheres as Advanced Anodes for Low-Cost, Safe, and High-Areal-Capacity Lithium-Ion Full Batteries. ACS Appl. Nano Mater. 2020. [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Huskinson, B.; Marshak, M.P.; Suh, C.; Er, S.; Gerhardt, M.R.; Galvin, C.J.; Chen, X.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. A metal-free organic-inorganic aqueous flow battery. Nature 2014, 505, 195–198. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef]
- Magasinski, A.; Dixon, P.; Hertzberg, B.; Kvit, A.; Ayala, J.; Yushin, G. High-performance lithium-ion anodes using a hierarchical bottom-up approach. Nat. Mater. 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, H.B.; Madhavi, S.; Hng, H.H.; Lou, X.W. Formation of Fe 2O 3 microboxes with hierarchical shell structures from metal-organic frameworks and their lithium storage properties. J. Am. Chem. Soc. 2012, 134, 17388–17391. [Google Scholar] [CrossRef]
- Han, D.; Zhang, J.; Weng, Z.; Kong, D.; Tao, Y.; Ding, F.; Ruan, D.; Yang, Q.H. Two-dimensional materials for lithium/sodium-ion capacitors. Mater. Today Energy 2019, 11, 30–45. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, N.; Cui, Y. Promises and challenges of nanomaterials for lithium-based rechargeable batteries. Nat. Energy 2016, 1. [Google Scholar] [CrossRef]
- Pramudita, J.C.; Sehrawat, D.; Goonetilleke, D.; Sharma, N. An Initial Review of the Status of Electrode Materials for Potassium-Ion Batteries. Adv. Energy Mater. 2017, 7, 1602911. [Google Scholar] [CrossRef]
- Schmuch, R.; Wagner, R.; Hörpel, G.; Placke, T.; Winter, M. Performance and cost of materials for lithium-based rechargeable automotive batteries. Nat. Energy 2018, 3, 267–278. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research development on sodium-ion batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhou, Z. MXene-based materials for electrochemical energy storage. J. Energy Chem. 2018, 27, 73–85. [Google Scholar] [CrossRef]
- Su, D.S.; Centi, G. A perspective on carbon materials for future energy application. J. Energy Chem. 2013, 22, 151–173. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, Y.; Lian, R.; Yang, D.; Zhang, D.; Meng, X.; Liu, Y.; Wei, Y.; Chen, G. Atomic insight into the structural transformation and anionic/cationic redox reactions of VS2 nanosheets in sodium-ion batteries. J. Mater. Chem. A 2018, 6, 15985–15992. [Google Scholar] [CrossRef]
- Xie, Y.; Dall’Agnese, Y.; Naguib, M.; Gogotsi, Y.; Barsoum, M.W.; Zhuang, H.L.; Kent, P.R.C. Prediction and characterization of mxene nanosheet anodes for non-lithium-ion batteries. ACS Nano 2014, 8, 9606–9615. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhao, M.Q.; Anasori, B.; Maleski, K.; Ren, C.E.; Li, J.; Byles, B.W.; Pomerantseva, E.; Wang, G.; Gogotsi, Y. Porous heterostructured MXene/carbon nanotube composite paper with high volumetric capacity for sodium-based energy storage devices. Nano Energy 2016, 26, 513–523. [Google Scholar] [CrossRef]
- Salavati, M.; Rabczuk, T. Application of highly stretchable and conductive two-dimensional 1T VS2 and VSe2 as anode materials for Li-, Na- and Ca-ion storage. Comput. Mater. Sci. 2019, 160, 360–367. [Google Scholar] [CrossRef]
- Wu, Y.; Nie, P.; Jiang, J.; Ding, B.; Dou, H.; Zhang, X. MoS 2 -Nanosheet-Decorated 2D Titanium Carbide (MXene) as High-Performance Anodes for Sodium-Ion Batteries. ChemElectroChem 2017, 4, 1560–1565. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, D.; Yang, D.; Wei, L.; Liu, B.; Wang, X.; Chen, G.; Wei, Y. Superior Mg2+ storage properties of VS2 nanosheets by using an APC-PP14Cl/THF electrolyte. Energy Storage Mater. 2019, 23, 749–756. [Google Scholar] [CrossRef]
- Mortazavi, B.; Makaremi, M.; Shahrokhi, M.; Raeisi, M.; Singh, C.V.; Rabczuk, T.; Pereira, L.F.C. Borophene hydride: A stiff 2D material with high thermal conductivity and attractive optical and electronic properties. Nanoscale 2018, 10, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Salavati, M.; Alajlan, N.; Rabczuk, T. Super-stretchability in two-dimensional RuCl3 and RuBr3 confirmed by first-principles simulations. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 113. [Google Scholar] [CrossRef]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, S.; Liu, H.; Li, Y.; Cui, G.; Li, Y. Graphdiyne for high capacity and long-life lithium storage. Nano Energy 2015, 11, 481–489. [Google Scholar] [CrossRef]
- Mortazavi, B.; Dianat, A.; Rahaman, O.; Cuniberti, G.; Rabczuk, T. Borophene as an anode material for Ca, Mg, Na or Li ion storage: A first-principle study. J. Power Sources 2016, 329, 456–461. [Google Scholar] [CrossRef]
- Li, L.; Zuo, Z.; Shang, H.; Wang, F.; Li, Y. In-situ constructing 3D graphdiyne as all-carbon binder for high-performance silicon anode. Nano Energy 2018, 53, 135–143. [Google Scholar] [CrossRef]
- Kuang, P.; Zhu, B.; Li, Y.; Liu, H.; Yu, J.; Fan, K. Graphdiyne: A superior carbon additive to boost the activity of water oxidation catalysts. Nanoscale Horiz. 2018, 3, 317–326. [Google Scholar] [CrossRef]
- Shang, H.; Zuo, Z.; Li, L.; Wang, F.; Liu, H.; Li, Y.; Li, Y. Ultrathin Graphdiyne Nanosheets Grown In Situ on Copper Nanowires and Their Performance as Lithium-Ion Battery Anodes. Angew. Chemie Int. Ed. 2017, 57, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Zuo, Z.; Yu, L.; Wang, F.; He, F.; Li, Y. Low-Temperature Growth of All-Carbon Graphdiyne on a Silicon Anode for High-Performance Lithium-Ion Batteries. Adv. Mater. 2018, 30, 1801459. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, R.; Toyoda, R.; Shiotsuki, R.; Fukui, N.; Wada, K.; Maeda, H.; Sakamoto, R.; Sasaki, S.; Masunaga, H.; Nagashio, K.; et al. Expansion of the Graphdiyne Family: A Triphenylene-Cored Analogue. ACS Appl. Mater. Interfaces 2019, 11, 2730–2733. [Google Scholar] [CrossRef]
- Matsuoka, R.; Sakamoto, R.; Hoshiko, K.; Sasaki, S.; Masunaga, H.; Nagashio, K.; Nishihara, H. Crystalline Graphdiyne Nanosheets Produced at a Gas/Liquid or Liquid/Liquid Interface. J. Am. Chem. Soc. 2017, 139, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.T.; Lee, S.; Park, S.; Lee, C.Y. Graphene chemiresistors modified with functionalized triphenylene for highly sensitive and selective detection of dimethyl methylphosphonate. RSC Adv. 2019, 9, 33976–33980. [Google Scholar] [CrossRef]
- Taghilou, H.; Fathi, D. Spin related transport in two pyrene and Triphenylene graphene nanodisks using NEGF method. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 101, 208–211. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shahrokhi, M.; Madjet, M.E.; Makaremi, M.; Ahzi, S.; Rabczuk, T. N-, P-, As-triphenylene-graphdiyne: Strong and stable 2D semiconductors with outstanding capacities as anodes for Li-ion batteries. Carbon 2019, 141, 291–303. [Google Scholar] [CrossRef]
- Srinivasu, K.; Ghosh, S.K. Graphyne and graphdiyne: Promising materials for nanoelectronics and energy storage applications. J. Phys. Chem. C 2012, 116, 5951–5956. [Google Scholar] [CrossRef]
- Li, X.; Wang, N.; He, J.; Yang, Z.; Tu, Z.; Zhao, F.; Wang, K.; Yi, Y.; Huang, C. Designing the efficient lithium diffusion and storage channels based on graphdiyne. Carbon N. Y. 2020, 162, 579–585. [Google Scholar] [CrossRef]
- Jia, Z.; Li, Y.; Zuo, Z.; Liu, H.; Huang, C.; Li, Y. Synthesis and Properties of 2D Carbon - Graphdiyne. Acc. Chem. Res. 2017, 50, 2470–2478. [Google Scholar] [CrossRef]
- Sun, C.; Searles, D.J. Lithium storage on graphdiyne predicted by DFT calculations. J. Phys. Chem. C 2012, 116, 26222–26226. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, H.; Guo, Y.; Li, Y.; Zhu, D. Architecture of graphdiyne nanoscale films. Chem. Commun. 2010, 46, 3256–3258. [Google Scholar] [CrossRef]
- Baughman, R.H.; Eckhardt, H.; Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp 2 and sp atoms. J. Chem. Phys. 1987, 87, 6687–6699. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Liu, H.; Li, Y. Graphdiyne and graphyne: From theoretical predictions to practical construction. Chem. Soc. Rev. 2014, 43, 2572–2586. [Google Scholar] [CrossRef]
- Long, M.; Tang, L.; Wang, D.; Li, Y.; Shuai, Z. Electronic structure and carrier mobility in graphdiyne sheet and nanoribbons: Theoretical predictions. ACS Nano 2011, 5, 2593–2600. [Google Scholar] [CrossRef]
- Pan, L.D.; Zhang, L.Z.; Song, B.Q.; Du, S.X.; Gao, H.J. Graphyne- and graphdiyne-based nanoribbons: Density functional theory calculations of electronic structures. Appl. Phys. Lett. 2011, 98, 173102. [Google Scholar] [CrossRef]
- Mortazavi, B.; Makaremi, M.; Shahrokhi, M.; Fan, Z.; Rabczuk, T. N-graphdiyne two-dimensional nanomaterials: Semiconductors with low thermal conductivity and high stretchability. Carbon N. Y. 2018, 137, 57–67. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shahrokhi, M.; Zhuang, X.; Rabczuk, T. Boron-graphdiyne: A superstretchable semiconductor with low thermal conductivity and ultrahigh capacity for Li, Na and Ca ion storage. J. Mater. Chem. A 2018, 6, 11022–11036. [Google Scholar] [CrossRef]
- Wang, N.; Li, X.; Tu, Z.; Zhao, F.; He, J.; Guan, Z.; Huang, C.; Yi, Y.; Li, Y. Synthesis and Electronic Structure of Boron-Graphdiyne with an sp-Hybridized Carbon Skeleton and Its Application in Sodium Storage. Angew. Chemie Int. Ed. 2018, 57, 3968–3973. [Google Scholar] [CrossRef]
- Lu, C.; Yang, Y.; Wang, J.; Fu, R.; Zhao, X.; Zhao, L.; Ming, Y.; Hu, Y.; Lin, H.; Tao, X.; et al. High-performance graphdiyne-based electrochemical actuators. Nat. Commun. 2018, 9, 752. [Google Scholar] [CrossRef]
- Zhao, Y.; Wan, J.; Yao, H.; Zhang, L.; Lin, K.; Wang, L.; Yang, N.; Liu, D.; Song, L.; Zhu, J.; et al. Few-layer graphdiyne doped with sp-hybridized nitrogen atoms at acetylenic sites for oxygen reduction electrocatalysis. Nat. Chem. 2018, 10, 924–931. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shahrokhi, M.; Madjet, M.E.; Hussain, T.; Zhuang, X.; Rabczuk, T. N-, B-, P-, Al-, As-, and Ga-graphdiyne/Graphyne Lattices: First-Principles Investigation of Mechanical, Optical and Electronic Properties. Available online: https://pubs.rsc.org/en/content/articlelanding/2019/tc/c9tc00082h/unauth#!divAbstract (accessed on 8 February 2019).
- Salavati, M.; Rabczuk, T. First-principles investigation of N-triphenylene-graphdiyne nanosheets as an anode material for Na, K, Mg and Ca storage. Comput. Mater. Sci. 2019, 169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Mortazavi, B.; Dianat, A.; Cuniberti, G.; Rabczuk, T. Application of silicene, germanene and stanene for Na or Li ion storage: A theoretical investigation. Electrochim. Acta 2016, 213, 865–870. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Scheffler, M.; Stampfl, C. Chapter 5 Theory of adsorption on metal substrates. Handb. Surf. Sci. 2000, 2, 285–356. [Google Scholar] [CrossRef]
- Aydinol, M.; Kohan, A.; Ceder, G.; Cho, K.; Joannopoulos, J. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 1354–1365. [Google Scholar] [CrossRef]
- Lee, H.; Jang, B.; Koo, J.; Park, M.; Lee, H.; Nam, J.; Kwon, Y. Graphdiyne as a high-capacity lithium ion battery anode material. Appl. Phys. Lett. 2013, 103, 263904. [Google Scholar] [CrossRef]
- Li, J.; Jiu, T.; Chen, S.; Liu, L.; Yao, Q.; Bi, F.; Zhao, C.; Wang, Z.; Zhao, M.; Zhang, G.; et al. Graphdiyne as a Host Active Material for Perovskite Solar Cell Application. Nano Lett. 2018, 18, 6941–6947. [Google Scholar] [CrossRef] [PubMed]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Pollak, E.; Geng, B.; Jeon, K.J.; Lucas, I.T.; Richardson, T.J.; Wang, F.; Kostecki, R. The interaction of Li+ with single-layer and few-layer graphene. Nano Lett. 2010, 10, 3386–3388. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hassanein, A. Kinetic Monte Carlo simulation of hydrogen diffusion on tungsten reconstructed (0 0 1) surface. Fusion Eng. Des. 2014, 89, 2545–2549. [Google Scholar] [CrossRef]
- Moon, J.; Lee, B.; Cho, M.; Cho, K. Ab initio and kinetic Monte Carlo simulation study of lithiation in crystalline and amorphous silicon. J. Power Sources 2014, 272, 1010–1017. [Google Scholar] [CrossRef]
- Zhong, K.; Hu, R.; Xu, G.; Yang, Y.; Zhang, J.M.; Huang, Z. Adsorption and ultrafast diffusion of lithium in bilayer graphene: Ab initio and kinetic Monte Carlo simulation study. Phys. Rev. B 2019, 99, 155403. [Google Scholar] [CrossRef]
- Hu, R.; Xu, G.; Yang, Y.; Zhang, J.M.; Zhong, K.; Huang, Z. Effect of stacking structure on lithium adsorption and diffusion in bilayer black phosphorene. Phys. Rev. B 2019, 100, 085422. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Li | Na | K | Ca | ||
|---|---|---|---|---|---|
| Most stable adsorption sites | S1 | EAds = −2.026 eV | EAds = −2.476 eV | EAds = −3.013 eV | EAds = −2.428 eV |
| LC1-Li = 3.280 Å | LC1-Na = 4.263 Å | LC1-K = 4.260 Å | LC1-Ca = 4.226 Å | ||
| LC2-Li = 3.279 Å | LC2-Na = 4.263 Å | LC2-K = 4.260Å | LC2-Ca = 4.226 Å | ||
| LZ = 0 Å | LZ = 0 Å | LZ = 0 Å | LZ = 0 Å | ||
| ΔQ = 0.995 |e| | ΔQ = 0.996 |e| | ΔQ = 0.850 |e| | ΔQ = 1.620 |e| | ||
| S2 | EAds = −1.703eV | EAds = −1.735eV | EAds = −2.368 eV | EAds = −1.804 eV | |
| LP1-Li = 2.558 Å | LP1-Na = 2.901 Å | LP1-K = 3.297 Å | LP1-Ca = 2.734 Å | ||
| LP2-Li = 2.559 Å | LP2-Na = 2.900 Å | LP2-K = 3.302 Å | LP2-Ca = 2.733 Å | ||
| LZ = 1.678 Å | LZ = 2.163 Å | LZ = 2.514 Å | LZ = 2.027 Å | ||
| ΔQ = 0.993 |e| | ΔQ = 0.991 |e| | ΔQ = 0.932 |e| | ΔQ = 1.510 |e| | ||
| S3 | EAds = −1.567eV | EAds = −1.794eV | EAds = −2.284 eV | EAds = −1.712 eV | |
| LC-Li = 2.505 Å | LC-Na = 3.255 Å | LC-K = 2.965 Å | LC-Ca = 2.408 Å | ||
| LP-Li = 2.482 Å | LP-Na = 2.819 Å | LP-K = 3.399 Å | LP-Ca = 2.882 Å | ||
| LZ = 1.024 Å | LZ = 0.216 Å | LZ = 2.397 Å | LZ = 1.819 Å | ||
| ΔQ = 0.994 |e| | ΔQ = 0.993 |e| | ΔQ = 0.912 |e| | ΔQ = 1.615 |e| | ||
| S4 | EAds = −1.551 eV | EAds = −1.793 eV | EAds = −2.513 eV | EAds = −1.560 eV | |
| LC-Li = 2.840 Å | LC-Na = 3.239 Å | LC-K = 3.729 Å | LC-Ca = 2.631 Å | ||
| LP-Li = 2.487 Å | LP-Na = 2.807 Å | LP-K = 3.214 Å | LP-Ca = 2.647 Å | ||
| LZ = 0 Å | LZ = 0 Å | LZ = 0 Å | LZ = 0 Å | ||
| ΔQ = 0.993 |e| | ΔQ = 0.992 |e| | ΔQ = 0.910 |e| | ΔQ = 1.625 |e| |
| Maximum Barrier Energy | ||
|---|---|---|
| Ions | Path 1 | Path 2 |
| Li | 0.75 | 0.75 |
| 0.44 | 0.48 | |
| 0.34 | 0.28 | |
| Na | 1.00 | 1.00 |
| 0.25 | 0.26 | |
| 0.32 | 0.14 | |
| K | 0.78 | 0.78 |
| 0.15 | 0.11 | |
| 0.28 | 0.08 | |
| Ca | 1.42 | 1.42 |
| 0.62 | 0.38 | |
| 1.29 | 0.11 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salavati, M.; Alajlan, N.; Rabczuk, T. Theoretical Prediction of P-Triphenylene-Graphdiyne as an Excellent Anode Material for Li, Na, K, Mg, and Ca Batteries. Appl. Sci. 2021, 11, 2308. https://doi.org/10.3390/app11052308
Salavati M, Alajlan N, Rabczuk T. Theoretical Prediction of P-Triphenylene-Graphdiyne as an Excellent Anode Material for Li, Na, K, Mg, and Ca Batteries. Applied Sciences. 2021; 11(5):2308. https://doi.org/10.3390/app11052308
Chicago/Turabian StyleSalavati, Mohammad, Naif Alajlan, and Timon Rabczuk. 2021. "Theoretical Prediction of P-Triphenylene-Graphdiyne as an Excellent Anode Material for Li, Na, K, Mg, and Ca Batteries" Applied Sciences 11, no. 5: 2308. https://doi.org/10.3390/app11052308
APA StyleSalavati, M., Alajlan, N., & Rabczuk, T. (2021). Theoretical Prediction of P-Triphenylene-Graphdiyne as an Excellent Anode Material for Li, Na, K, Mg, and Ca Batteries. Applied Sciences, 11(5), 2308. https://doi.org/10.3390/app11052308