
Biological Evaluation of 4-(1H-triazol-1-yl)benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antioxidant Activity Investigation
2.2. Computation of Antioxidant Descriptors
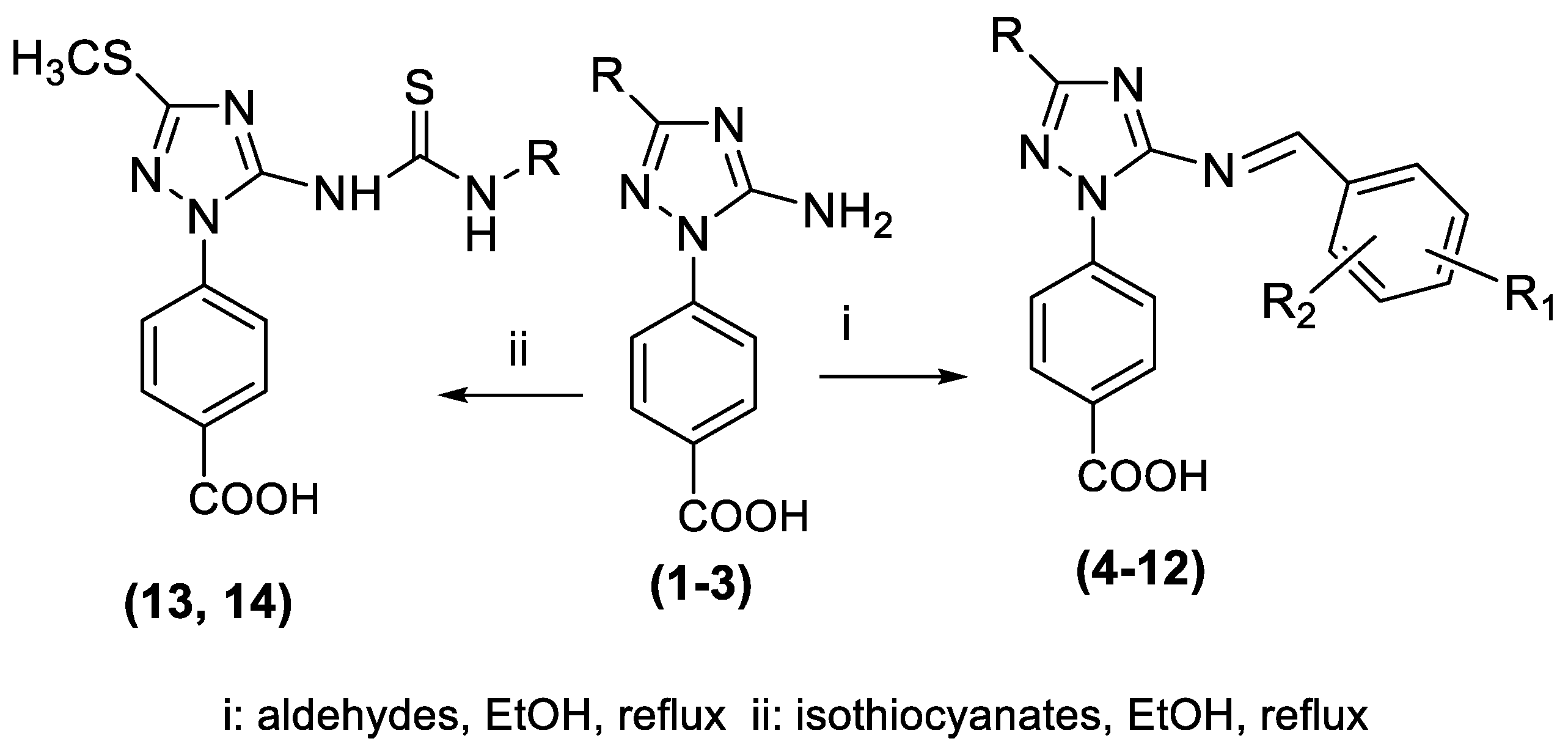
3. Results and Discussion
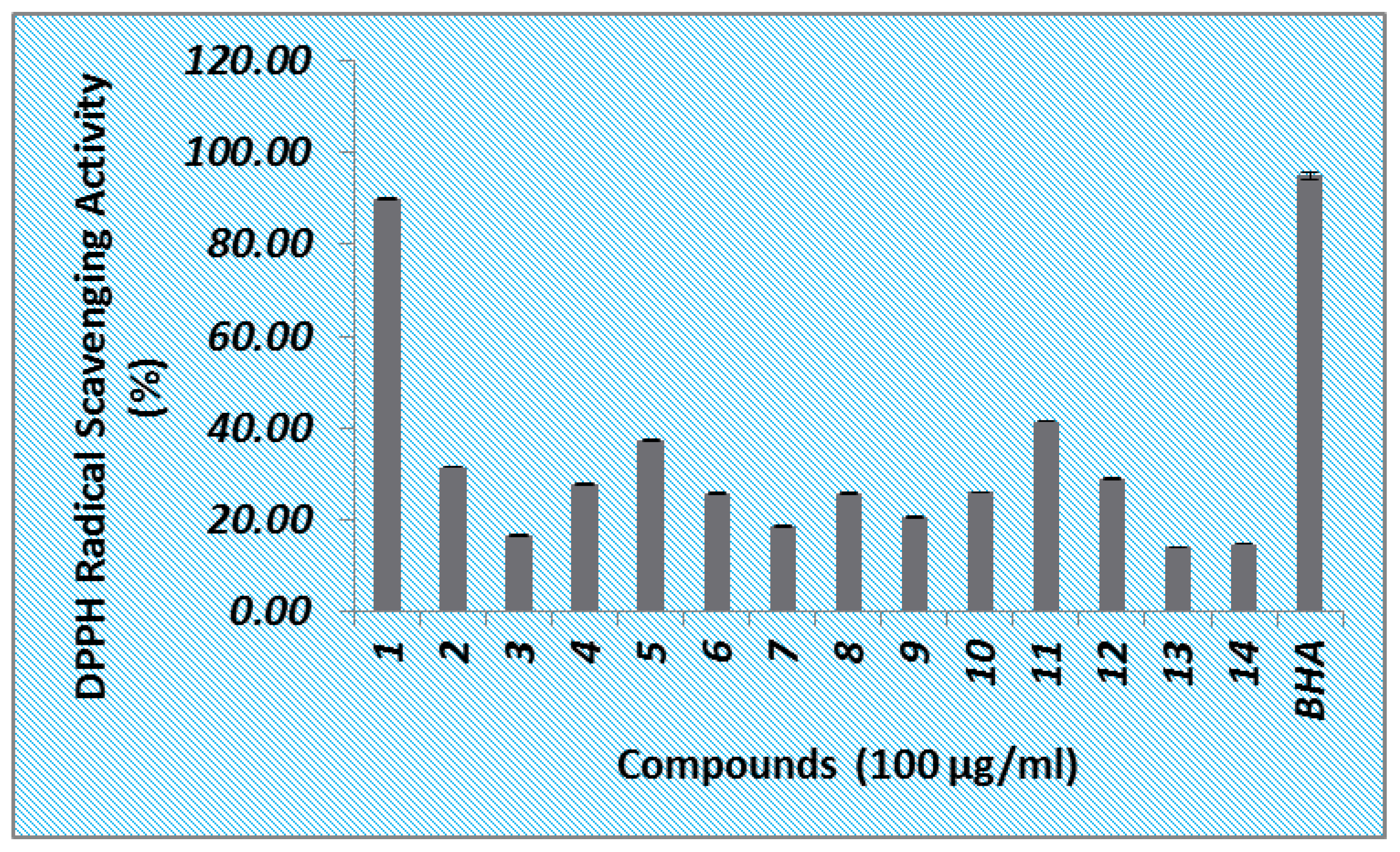
3.1. DPPH Radical Scavenging Activity
3.2. Reducing Power Ability
3.3. Ferric Reducing Power Activity
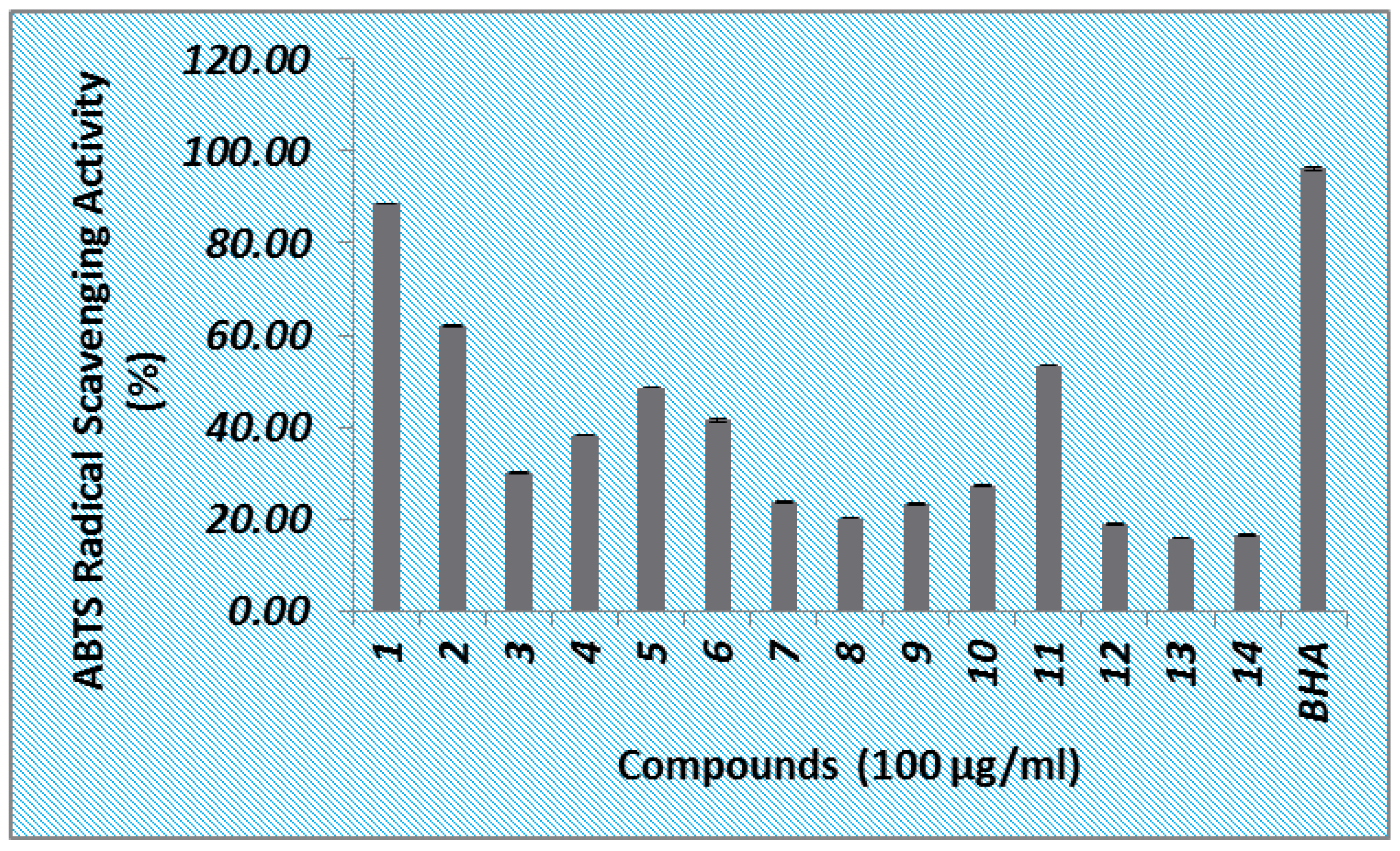
3.4. ABTS Radical Scavenging Activity
3.5. DFT Study
3.5.1. Geometry Optimization
3.5.2. Global Reactivity Descriptors
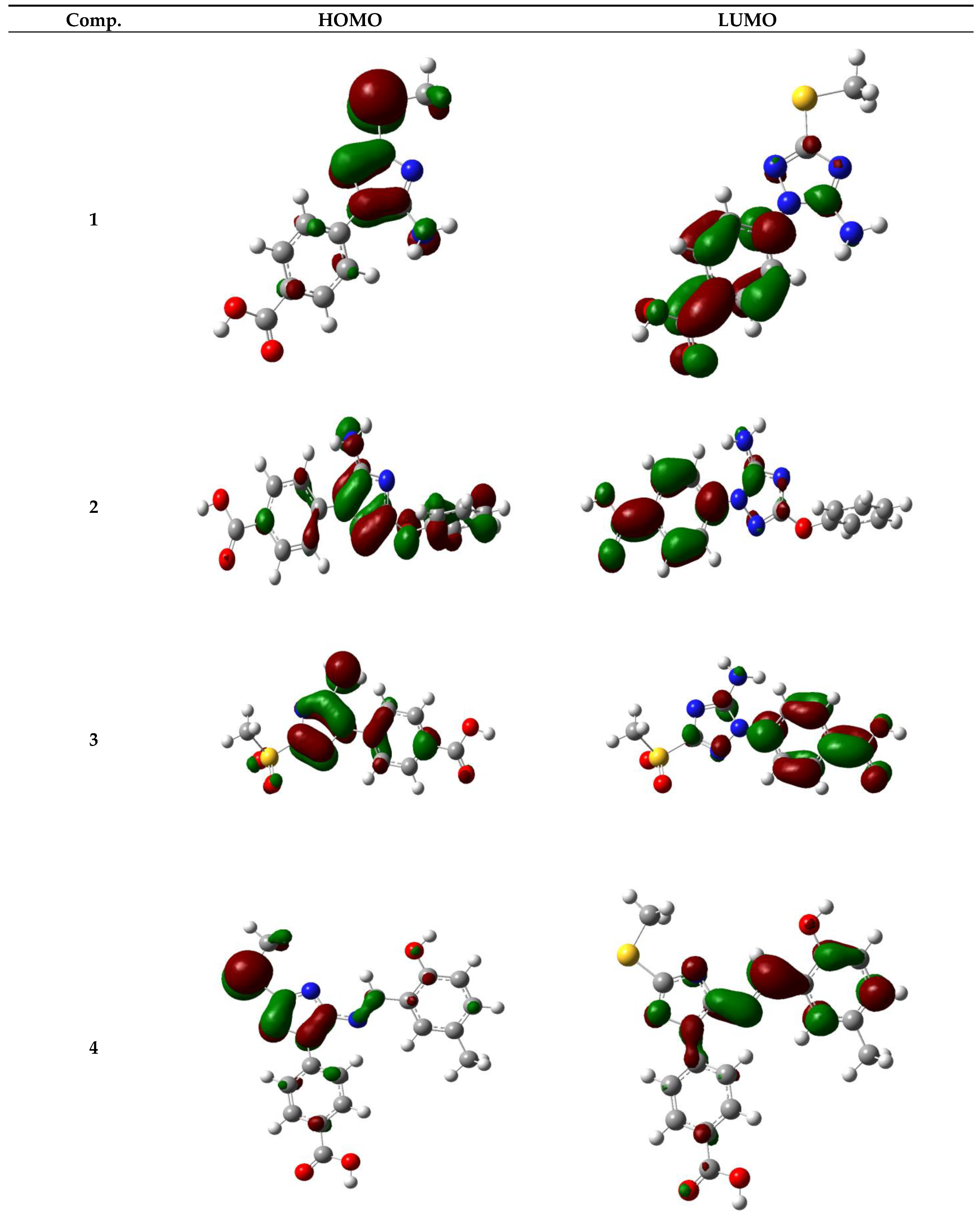
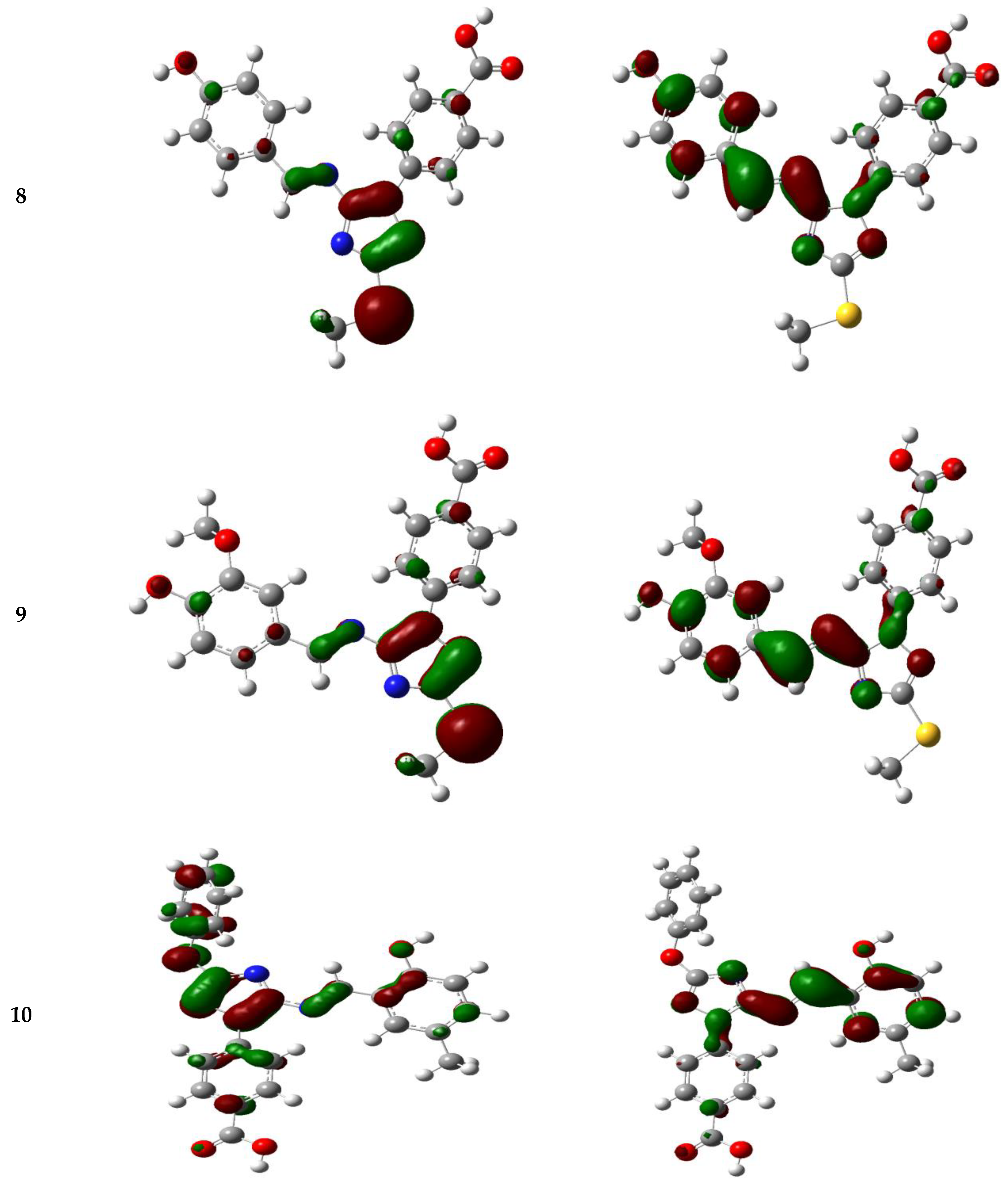
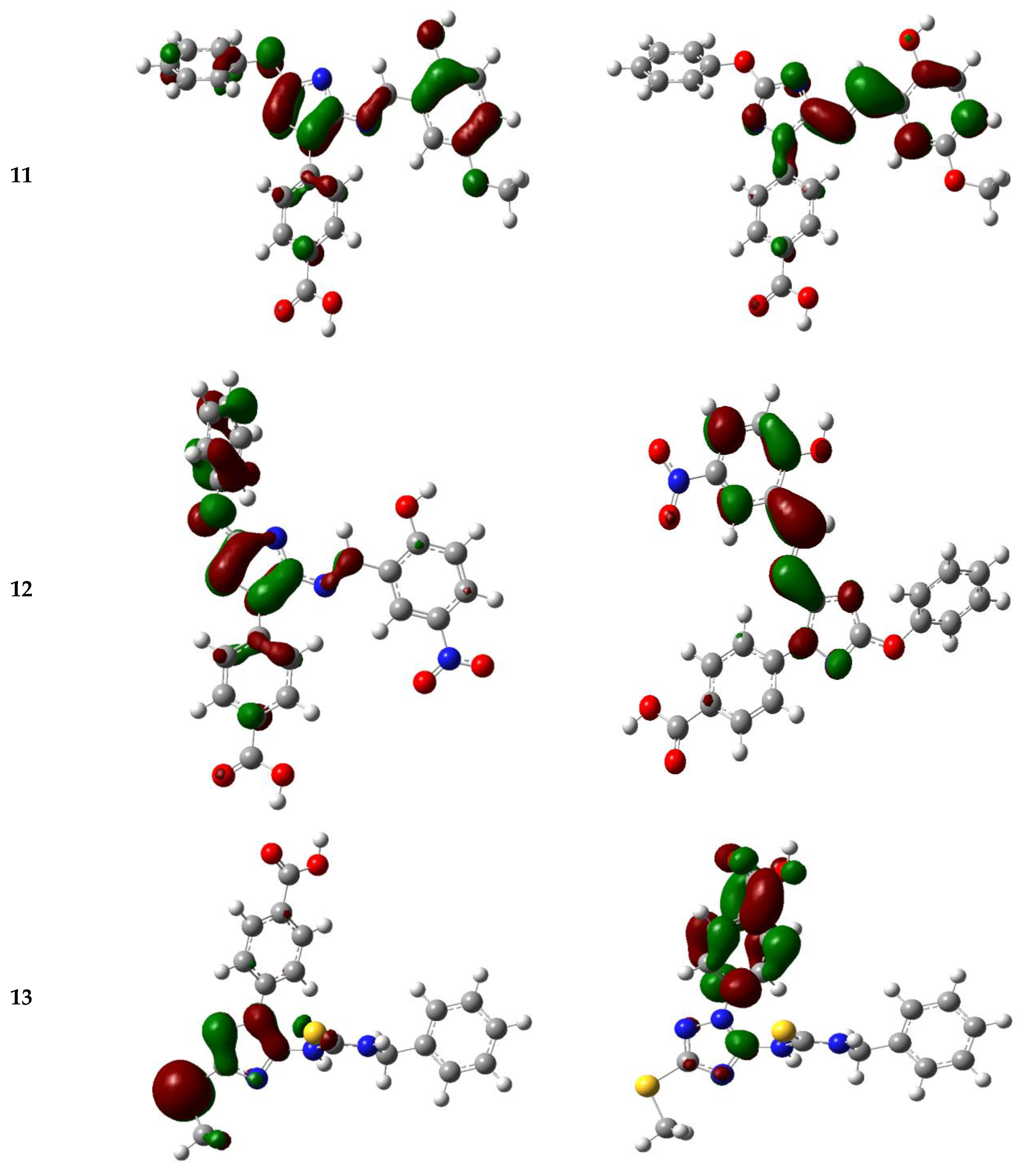
3.5.3. Frontier Molecular Orbitals
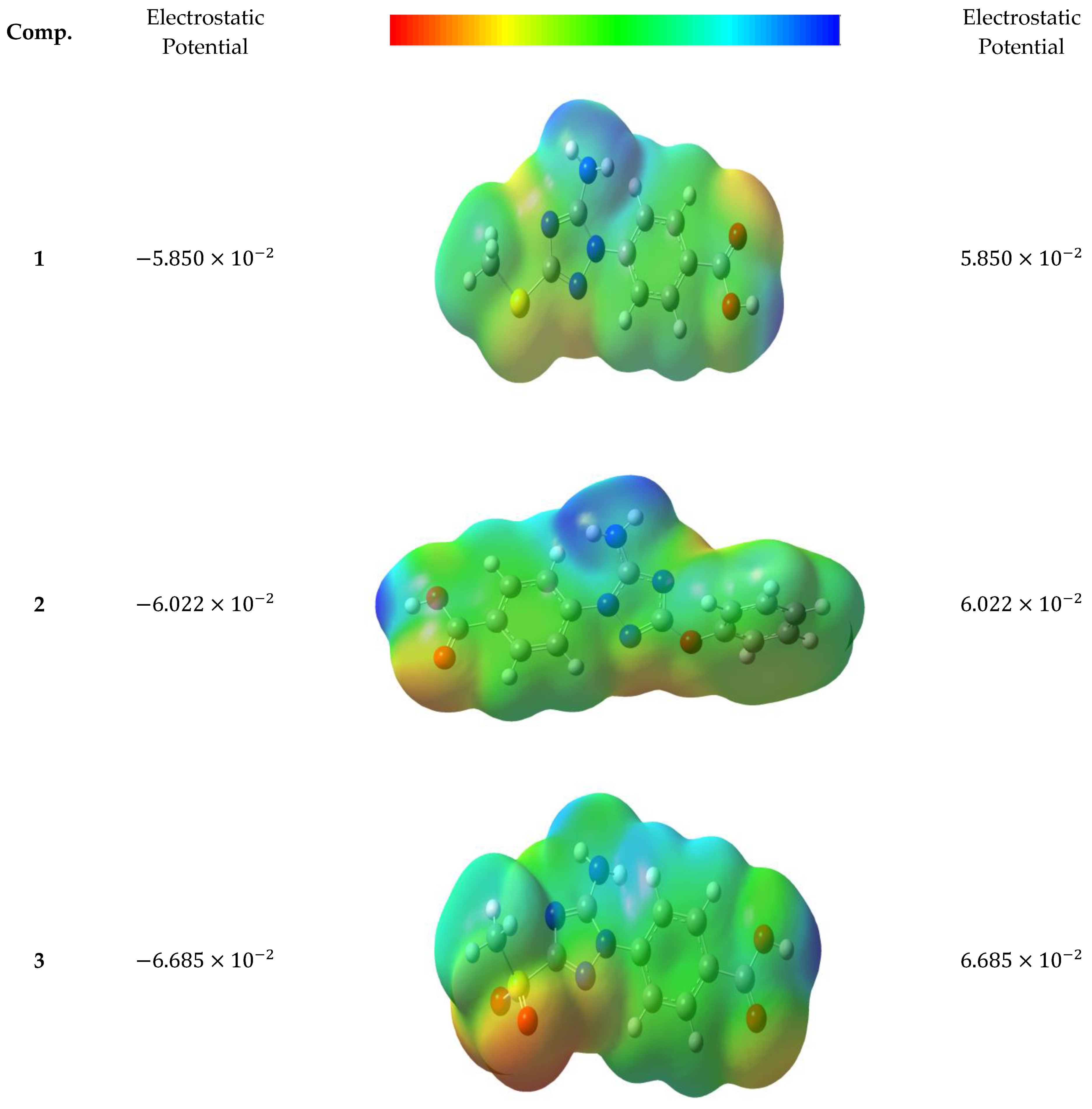
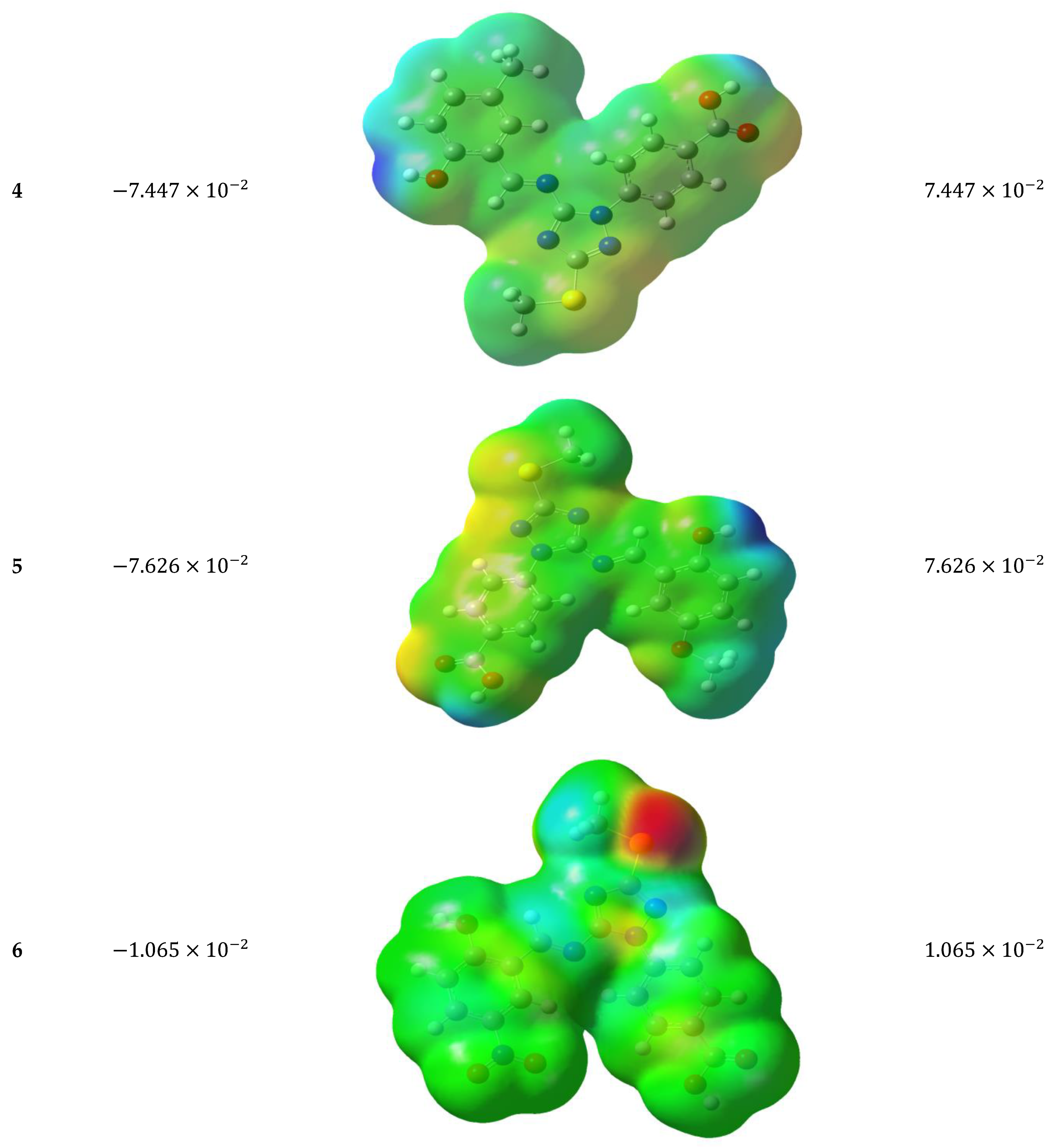
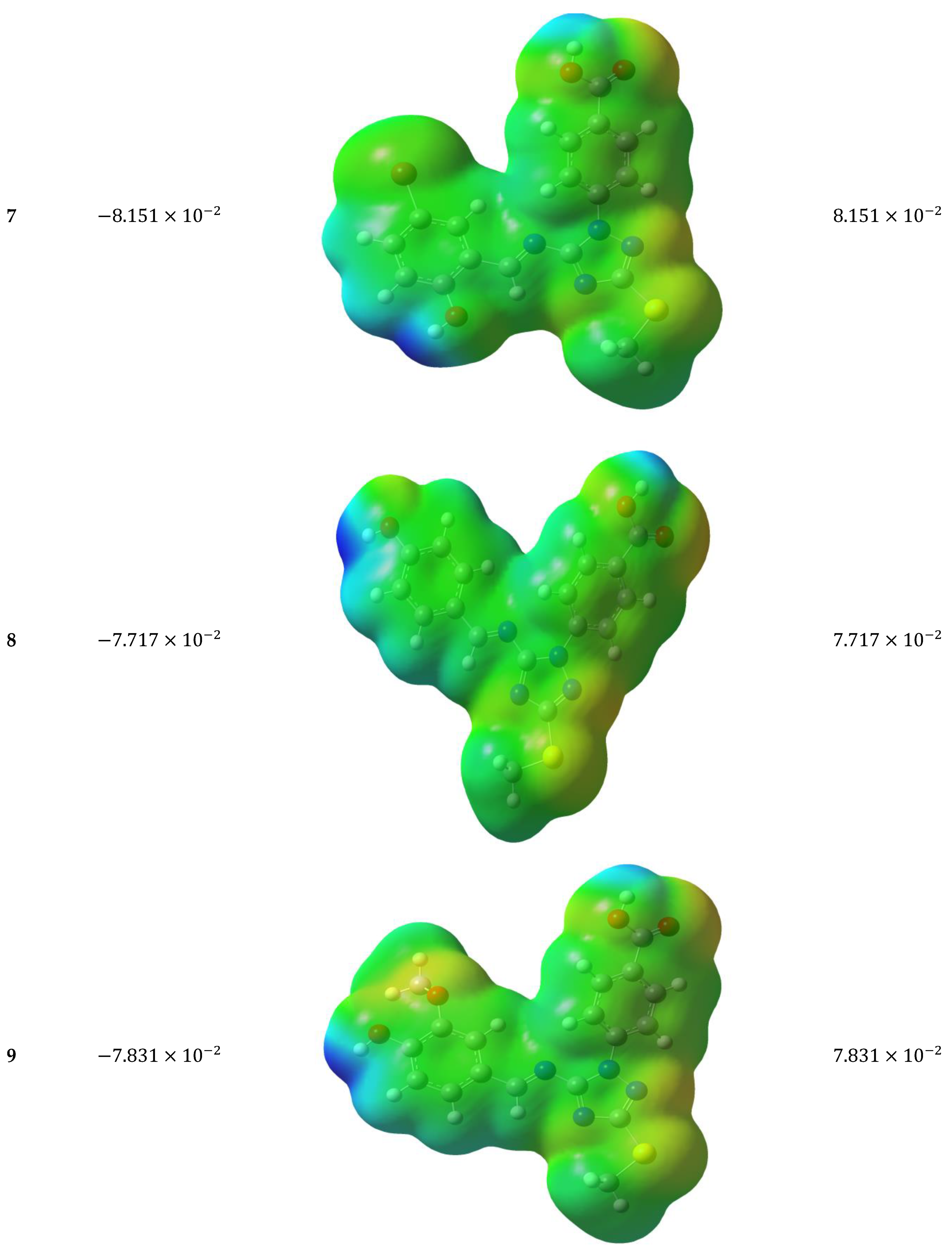
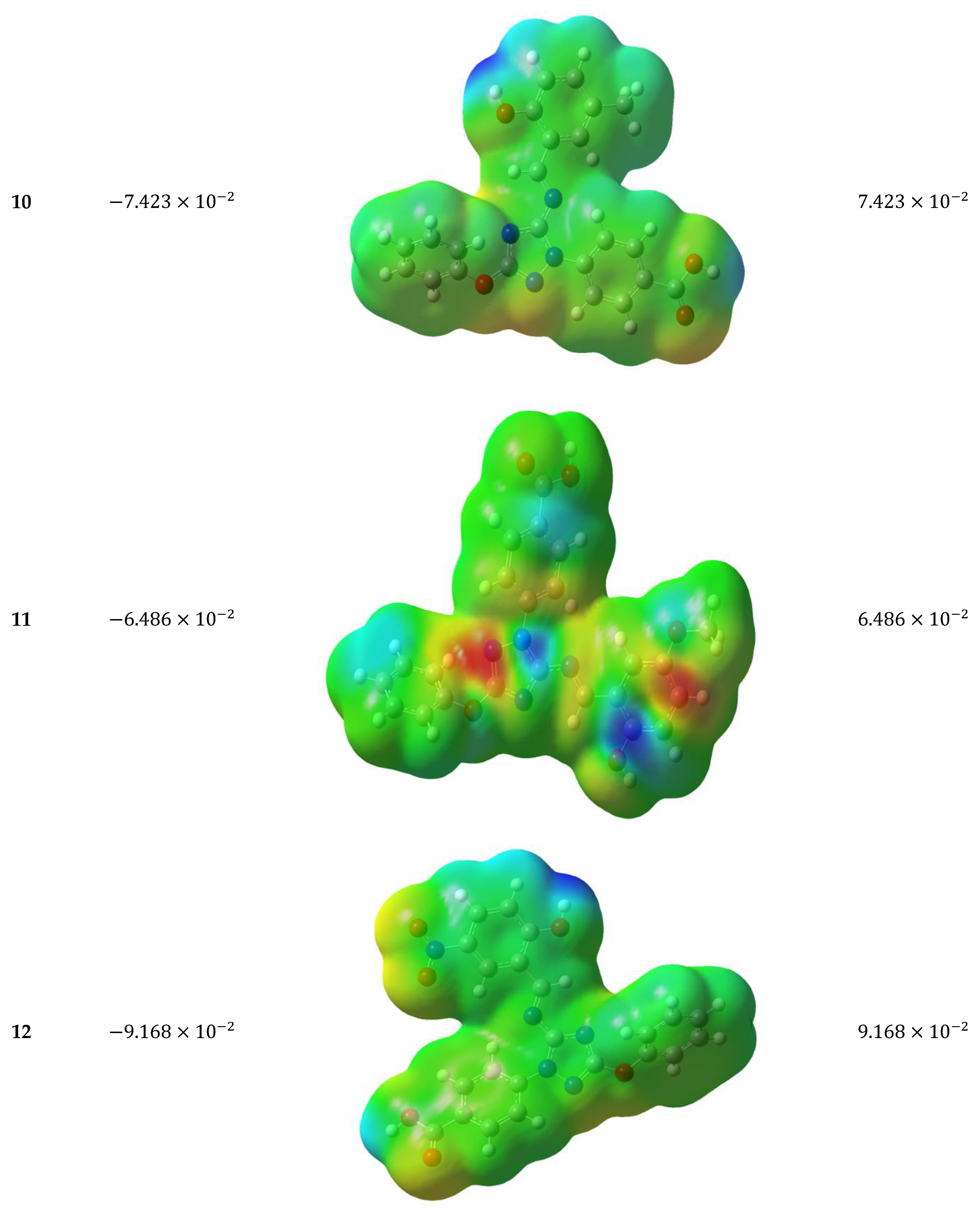
3.5.4. Molecular Electrostatic Potential (MEP)
3.5.5. Antioxidant Mechanisms of the Target Molecules 1–14
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kharb, R.; Sharma, P.C.; Shahar, M.Y. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Al-Salahi, R. An overview of triazoloquinazolines: Pharmacological significance and recent developments. Bioorg. Chem. 2021, 115, 10526. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Kaplancikli, Z.A.; Kiliç, F.S.; Erol, K. The synthesis of some triazolylphenothiazine derivatives and their antidepressant and anxiolytic activities. Boll. Chim. Farm. 2002, 141, 192–196. [Google Scholar]
- Luszczki, J.J.; Plech, T.; Wujec, M. Influence of 5-(3-chlorophenyl)-4-(4-methylphenyl)-2, 4-dihydro-3H-1, 2, 4-triazole-3-thione on the anticonvulsant action of 4 classical antiepileptic drugs in the mouse maximal electroshock-induced seizure model. Pharmacol. Rep. 2012, 64, 970–978. [Google Scholar] [CrossRef]
- Song, M.-X.; Deng, X.-Q. Recent developments on triazole nucleus in anticonvulsant compounds: A review. J. Enzym. Inhib. Med. Chem. 2018, 33, 453–478. [Google Scholar] [CrossRef] [Green Version]
- Veloso, R.V.; Shamim, A.; Lamarrey, Y.; Stefani, H.A.; Sciani, J.M. Antioxidant and anti-sickling activity of glucal-based triazoles compounds—An in vitro and in silico study. Bioorg. Chem. 2021, 109, 104709. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Anouar, E.-H.; Marzouk, M.; Taie, H.A.A.; Abuelizz, H.A. Screening and evaluation of antioxidant activity of some 1, 2, 4-triazolo [1, 5-a] quinazoline derivatives. Future Med. Chem. 2018, 10, 379–390. [Google Scholar] [CrossRef]
- Saadeh, H.A.; Mosleh, I.M.; Al-Bakri, A.G.; Mubarak, M.S. Synthesis and antimicrobial activity of new 1, 2, 4-triazole-3-thiol metronidazole derivatives. Monatshefte Chem. 2010, 141, 471–478. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Marzouk, M.; Awad, G.; Al-Omar, M.; Ezzeldin, E. Antimicrobial activity of newly synthesized methylsulfanyl- triazoloquinazoline derivatives. J. Pharm. Pharmacol. 2013, 65, 790–797. [Google Scholar] [CrossRef]
- Cao, X.; Wang, W.; Wang, S.; Bao, L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017, 139, 718–725. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Al-Swaidan, I.; Al-Omar, M.; Marzouk, M. Antiviral activity of 2-phenoxy-[1,2,4]triazolo [1,5-α]quinazoline derivatives. Life Sci. J. 2013, 10, 2164–2169. [Google Scholar]
- Sun, L.; Huang, T.; Dick, A.; Meuser, M.E.; Zalloum, W.A.; Chen, C.-H.; Ding, X.; Gao, P.; Cocklin, S.; Lee, K.-H. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1, 2, 3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur. J. Med. Chem. 2020, 190, 112085. [Google Scholar] [CrossRef]
- Tozkoparan, B.; Küpeli, E.; Yeşilada, E.; Ertan, M. Preparation of 5-aryl-3-alkylthio-l, 2, 4-triazoles and corresponding sulfones with anti-inflammatory-analgesic activity. Bioorg. Med. Chem. 2007, 15, 1808–1814. [Google Scholar] [CrossRef]
- Ankali, K.N.; Rangaswamy, J.; Shalavadi, M.; Naik, N.; naik Krishnamurthy, G. Synthesis and molecular docking of novel 1, 3-thiazole derived 1, 2, 3-triazoles and in vivo biological evaluation for their anti anxiety and anti inflammatory activity. J. Mol. Struct. 2021, 1236, 130357. [Google Scholar] [CrossRef]
- Iqbal, A.K.M.; Khan, A.Y.; Kalashetti, M.B.; Belavagi, N.S.; Gong, Y.-D.; Khazi, I.A.M. Synthesis, hypoglycemic and hypolipidemic activities of novel thiazolidinedione derivatives containing thiazole/triazole/oxadiazole ring. Eur. J. Med. Chem. 2012, 53, 308–315. [Google Scholar] [CrossRef]
- Luzina, E.L.; Popov, A.V. Anticancer activity of N-bis (trifluoromethyl) alkyl-N′-(polychlorophenyl) and N′-(1, 2, 4-triazolyl) ureas. Eur. J. Med. Chem. 2010, 45, 5507–5512. [Google Scholar] [CrossRef]
- El-Sherief, H.A.M.; Youssif, B.G.M.; Bukhari, S.N.A.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Novel 1, 2, 4-triazole derivatives as potential anticancer agents: Design, synthesis, molecular docking and mechanistic studies. Bioorg. Chem. 2018, 76, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.K.; Mishra, P. Pharmacological screening of some newly synthesized triazoles for H 1 receptor antagonist activity. Med. Chem. Res. 2017, 26, 2260–2271. [Google Scholar] [CrossRef]
- Al-Salahi, R.A.; Alswaidan, I.A.; Marzouk, M.S.; El-Eraky, W.; Saleh, D. Antihistamine activity of 1,2,4-triazolo[1,5-a]quinazolines. Asian J. Chem. 2014, 26, 8617–8619. [Google Scholar] [CrossRef]
- Chai, B.; Qian, X.; Cao, S.; Liu, H.; Song, G. Synthesis and insecticidal activity of 1, 2, 4-triazole derivatives. Arkivoc 2003, 2, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.S.; Misra, H.K. Synthesis and pesticidal activities of some new substituted 1, 2, 4-triazoles and their derivatives. Agric. Biol. Chem. 1980, 44, 1009–1013. [Google Scholar] [CrossRef]
- Parmar, S.S.; Gupta, A.K.; Singh, H.H.; Gupta, T.K. Benzimidazolyl-1, 2, 4-(H)-triazoles as central nervous system depressants. J. Med. Chem. 1972, 15, 999–1000. [Google Scholar] [CrossRef]
- Perekhoda, L.; Georgiyants, V.; Yeromina, H.; Drapak, I.; Lubenets, V.; Ieromina, Z.; Sych, I.; Severina, H.; Demchenko, A. The synthesis and in silico antihypertensive activity prognosis of new mannich bases containing the 1, 2, 4-triazole moiety. Chem. Chem. Technol. 2020, 2, 214–220. [Google Scholar] [CrossRef]
- Al-Salahi, R.; El-Tahir, K.-E.; Alswaidan, I.; Lolak, N.; Hamidaddin, M.; Marzouk, M. Biological effects of a new set 1,2,4-triazolo[1,5-a]quinazolines on heart rate and blood pressure. Chem. Cent. J. 2014, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Küçükgüzel, Ş.G.; Çıkla-Süzgün, P. Recent advances bioactive 1, 2, 4-triazole-3-thiones. Eur. J. Med. Chem. 2015, 97, 830–870. [Google Scholar] [CrossRef]
- Rajavelu, K.; Subaraja, M.; Rajakumar, P. Synthesis, optical properties, and antioxidant and anticancer activity of benzoheterazole dendrimers with triazole bridging unit. New J. Chem. 2018, 42, 3282–3292. [Google Scholar] [CrossRef]
- Settypalli, T.; Chunduri, V.R.; Maddineni, A.K.; Begari, N.; Allagadda, R.; Kotha, P.; Chippada, A.R. Design, synthesis, in silico docking studies and biological evaluation of novel quinoxaline-hydrazide hydrazone-1, 2, 3-triazole hybrids as α-glucosidase inhibitors and antioxidants. New J. Chem. 2019, 43, 15435–15452. [Google Scholar] [CrossRef]
- Savegnago, L.; do Sacramento, M.; Brod, L.M.P.; Fronza, M.G.; Seus, N.; Lenardao, E.J.; Paixão, M.W.; Alves, D. Phenylselanyl-1 H-1, 2, 3-triazole-4-carbonitriles: Synthesis, antioxidant properties and use as precursors to highly functionalized tetrazoles. RSC Adv. 2016, 6, 8021–8031. [Google Scholar] [CrossRef]
- Beyzaei, H.; Kudeyani, M.G.; Delarami, H.S.; Aryan, R. Synthesis, antimicrobial and antioxidant evaluation, and molecular docking study of 4, 5-disubstituted 1, 2, 4-triazole-3-thiones. J. Mol. Struct. 2020, 1215, 128273. [Google Scholar] [CrossRef]
- Yadav, S.K.; Yadav, R.K.; Yadava, U. Computational investigations and molecular dynamics simulations envisioned for potent antioxidant and anticancer drugs using indole-chalcone-triazole hybrids. DNA Repair 2020, 86, 102765. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Anouar, E.H.; Marzouk, M.; Hasan, M.H.; Saleh, S.R.; Ahudhaif, A.; Alburikan, K.A.; Al-Salahi, R. Evaluation of cytotoxic and tyrosinase inhibitory activities of 2-phenoxy (thiomethyl) pyridotriazolopyrimidines: In vitro and molecular docking studies. Anti-Cancer Agents Med. Chem. 2020, 20, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Taie, H.A.A.; Marzouk, M.; Al-Salahi, R. Synthesis and antioxidant activity of 2-methylthio-pyrido [3, 2-e][1,2,4] triazolo [1,5-a] pyrimidines. Open Chem. 2019, 17, 823–830. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Marzouk, M.; Ashour, A.E.; Alswaidan, I. Synthesis and antitumor activity of 1,2,4-triazolo[1,5-a]quinazolines. Asian J. Chem. 2014, 26, 173–2176. [Google Scholar] [CrossRef]
- Abuelizz, H.A.; Awad, H.M.; Marzouk, M.; Nasr, F.A.; Alqahtani, A.S.; Bakheit, A.H.; Naglah, A.M.; Al-Salahi, R. Synthesis and biological evaluation of 4-(1H-1,2,4-triazol-1-yl)benzoic acid hybrids as anticancer agents. RSC Adv. 2019, 9, 19065–19074. [Google Scholar] [CrossRef] [Green Version]
- Abuelizz, H.A.; Anouar, E.; Marzouk, M.; Taie, H.A.A.; Ahudhaif, A.; Al-Salahi, R. DFT study and radical scavenging activity of 2-phenoxypyridotriazolo pyrimidines by DPPH, ABTS, FRAP and reducing power capacity. Chem. Pap. 2020, 74, 2893–2899. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Taie, H.A.A.; Bakheit, A.H.; Marzouk, M.; Almehizia, A.A.; Herqash, R.; Abuelizz, H.A. Antioxidant activities and molecular docking of 2-thioxobenzo[g]quinazoline derivatives. Pharmacol. Rep. 2019, 71, 695–700. [Google Scholar] [CrossRef]
- Almehizia, A.A.; Abuelizz, H.A.; Taie, H.A.A.; ElHassane, A.; Marzouk, M.; Al-Salahi, R. Investigation the antioxidant activity of benzo[g]triazoloquinazolines correlated with a DFT study. Saudi Pharm. J. 2019, 27, 33–137. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Taie, H.A.A.; Bakheit, A.H.; Mostafa, G.A.E.; Marzouk, M.; Rashid, H.; Al-Salahi, R. Investigation of 4-Hydrazinobenzoic Acid Derivatives for Their Antioxidant Activity: In Vitro Screening and DFT Study. ACS Omega 2021, 6, 31993–32004. [Google Scholar] [CrossRef]
- Ruscic, B. Active thermochemical tables: Sequential bond dissociation enthalpies of methane, ethane, and methanol and the related thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Li, J. Theoretical study of the radical scavenging activity of Shikonin and its derivatives. Chin. J. Chem. 2012, 30, 84–90. [Google Scholar] [CrossRef]
- Mikulski, D.; Eder, K.; Molski, M. Quantum-chemical study on relationship between structure and antioxidant properties of hepatoprotective compounds occurring in Cynara scolymus and Silybum marianum. J. Theor. Comput. Chem. 2014, 13, 1450004. [Google Scholar] [CrossRef]
- Alisi, I.O.; Uzairu, A.; Abechi, S.E. Free radical scavenging mechanism of 1, 3, 4-oxadiazole derivatives: Thermodynamics of O–H and N–H bond cleavage. Heliyon 2020, 6, e03683. [Google Scholar] [CrossRef]
- Nenadis, N.; Tsimidou, M.Z. Contribution of DFT computed molecular descriptors in the study of radical scavenging activity trend of natural hydroxybenzaldehydes and corresponding acids. Food Res. Int. 2012, 48, 538–543. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the electron and the proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Bizarro, M.M.; Cabral, B.J.C.; Dos Santos, R.M.B.; Simões, J.A.M. Substituent effects on the O–H bond dissociation enthalpies in phenolic compounds: Agreements and controversies+ erratum. Pure Appl. Chem. 1999, 71, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Rimarčík, J.; Lukeš, V.; Klein, E.; Ilčin, M. Study of the solvent effect on the enthalpies of homolytic and heterolytic N–H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J. Mol. Struct. 2010, 952, 25–30. [Google Scholar] [CrossRef]
- Farrokhnia, M. Density functional theory studies on the antioxidant mechanism and electronic properties of some bioactive marine meroterpenoids: Sargahydroquionic acid and sargachromanol. ACS Omega 2020, 5, 20382–20390. [Google Scholar] [CrossRef]
- El Bahnasawy, R.M.; El Shereafy, E.; Kashar, T.I. Thermal and temperature dependence of electrical conductivity studies on Zn, Cd and Hg hydrazone complexes. J. Therm. Anal. 1993, 39, 65–74. [Google Scholar] [CrossRef]
- Nam, P.-C.; Nguyen, M.T.; Chandra, A.K. The c-h and α (c-x) bond dissociation enthalpies of toluene, c6h5-ch2x (x = f, cl), and their substituted derivatives: A dft study. J. Phys. Chem. A 2005, 109, 10342–10347. [Google Scholar] [CrossRef]
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.T.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.E.; Pohjamo, S.P.; Holmbom, B.R. Antioxidant activity of knotwood extractives and phenolic compounds of selected tree species. J. Agric. Food Chem. 2003, 51, 7600–7606. [Google Scholar] [CrossRef]
- Pietarinen, S.P.; Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Holmbom, B.R. Knotwood and bark extracts: Strong antioxidants from waste materials. J. Wood Sci. 2006, 52, 436–444. [Google Scholar] [CrossRef]
- Vo, Q.V.; Nam, P.C.; Van Bay, M.; Thong, N.M.; Cuong, N.D.; Mechler, A. Density functional theory study of the role of benzylic hydrogen atoms in the antioxidant properties of lignans. Sci. Rep. 2018, 8, 1–10. [Google Scholar]
- Pérez-González, A.; Galano, A. On the •OH and •OOH scavenging activity of 3-methyl-1-pyridin-2-yl-5-pyrazolone: Comparisons with its parent compound, edaravone. Int. J. Quantum Chem. 2012, 112, 3441–3448. [Google Scholar] [CrossRef]
- Li, M.; Liu, W.; Peng, C.; Ren, Q.; Lu, W.; Deng, W. A DFT study on reaction of eupatilin with hydroxyl radical in solution. Int. J. Quantum Chem. 2013, 113, 966–974. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 (1) | 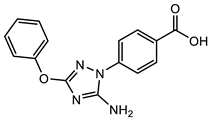 (2) | 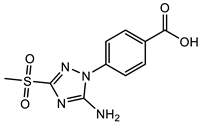 (3) |
 (4) | 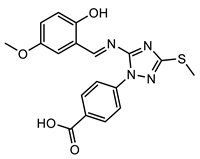 (5) | 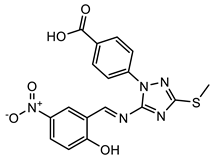 (6) |
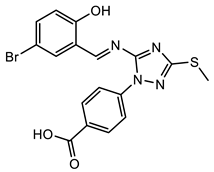 (7) |  (8) |  (9) |
 (10) |  (11) | 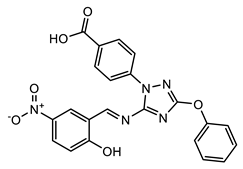 (12) |
 (13) | 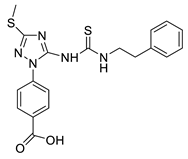 (14) |
| Compound | Reducing Power (Absorbance at 700 nm) |
|---|---|
| 1 | 1.43 ± 0.007 |
| 2 | 0.270 ± 0.004 |
| 3 | 0.31 ± 0.007 |
| 4 | 0.36 ± 0.019 |
| 5 | 0.37 ± 0.019 |
| 6 | 0.77 ± 0.014 |
| 7 | 0.16 ± 0.012 |
| 8 | 0.34 ± 0.020 |
| 9 | 0.58 ± 0.013 |
| 10 | 0.272 ± 0.006 |
| 11 | 0.21 ± 0.006 |
| 12 | 0.310 ± 0.010 |
| 13 | 0.37 ± 0.006 |
| 14 | 0.41 ± 0.008 |
| BHA | 1.76 ± 0.062 |
| Compound | FRAP Assay (μmol Trolox/100 g) |
|---|---|
| 1 | 3567 ± 14.84 |
| 2 | 953 ± 12.50 |
| 3 | 882 ± 7.09 |
| 4 | 462 ± 18.18 |
| 5 | 1482 ± 8.19 |
| 6 | 681 ± 9.07 |
| 7 | 385 ± 6.56 |
| 8 | 315 ± 7.37 |
| 9 | 1474 ± 11.06 |
| 10 | 542 ± 10.60 |
| 11 | 2093 ± 6.08 |
| 12 | 564 ± 12.86 |
| 13 | 520 ± 11.50 |
| 14 | 367 ± 9.71 |
| Comp. | εHOMO | εLUMO | Gap (kcal) | IP | EA | χ | μ | η | S | ω |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −137.818 | −48.098 | 89.72 | 137.818 | 48.098 | 92.958 | −92.958 | 44.86 | 0.022 | 193,821.2 |
| 2 | −146.534 | −39.087 | 107.447 | 146.534 | 39.087 | 92.81 | −92.81 | 53.723 | 0.019 | 231,380.5 |
| 3 | −165.61 | −56.218 | 109.392 | 165.61 | 56.218 | 110.914 | −110.914 | 54.696 | 0.018 | 336,431.6 |
| 4 | −136.092 | −60.491 | 75.601 | 136.092 | 60.491 | 98.292 | −98.292 | 37.801 | 0.026 | 182,600.3 |
| 5 | −136.356 | −61.928 | 74.428 | 136.356 | 61.928 | 99.142 | −99.142 | 37.214 | 0.027 | 182,889.7 |
| 6 | −141.953 | −71.378 | 70.575 | 141.953 | 71.378 | 106.666 | −106.666 | 35.287 | 0.028 | 200,742.4 |
| 7 | −138.991 | −65.781 | 73.21 | 138.991 | 65.781 | 102.386 | −102.386 | 36.605 | 0.027 | 191,864.4 |
| 8 | −137.052 | −59.638 | 77.415 | 137.052 | 59.638 | 98.345 | −98.345 | 38.707 | 0.026 | 187,183.4 |
| 9 | −136.632 | −59.374 | 77.258 | 136.632 | 59.374 | 98.003 | −98.003 | 38.629 | 0.026 | 185,507.1 |
| 10 | −142.229 | −61.162 | 81.067 | 142.229 | 61.162 | 101.696 | −101.696 | 40.533 | 0.025 | 209,598.7 |
| 11 | −137.353 | −56.488 | 80.866 | 137.353 | 56.488 | 96.921 | −96.921 | 40.433 | 0.025 | 189,905.3 |
| 12 | −144.789 | −67.927 | 76.862 | 144.789 | 67.927 | 106.358 | −106.358 | 38.431 | 0.026 | 217,368 |
| 13 | −136.977 | −47.326 | 89.651 | 136.977 | 47.326 | 92.152 | −92.152 | 44.825 | 0.022 | 190,326.7 |
| 14 | −142.957 | −38.052 | 104.905 | 142.957 | 38.052 | 90.504 | −90.504 | 52.453 | 0.019 | 214,821 |
| BHA | −129.76 | −8.555 | 121.206 | 5.627 | 8.555 | 69.159 | −69.159 | 60.603 | 8.786 | 39.457 |
| Comp. | BDE | AIP | PDE | PA | ETE | Dipole Moment | Polarizability (α) |
|---|---|---|---|---|---|---|---|
| 1 | 82.94 | 170.52 | 228.33 | 332.34 | 66.52 | 2.95 | 189.69 |
| 2 | 89.21 | 168.78 | 236.35 | 343.04 | 62.1 | 4.65 | 207.84 |
| 3 | 92.67 | 194.35 | 214.24 | 330.71 | 77.88 | 8.19 | 189.52 |
| 4 | 81.21 | 164.38 | 232.74 | 319.59 | 77.54 | 8.3 | 319.35 |
| 5 | 80.32 | 164.61 | 231.63 | 332.77 | 63.46 | 9.37 | 311.64 |
| 6 | 82.7 | 172.24 | 226.38 | 305.49 | 93.13 | 8.72 | 324.28 |
| 7 | 81.14 | 167.79 | 229.27 | 314.04 | 83.02 | 7.85 | 326.89 |
| 8 | 81.51 | 165.05 | 232.38 | 316.55 | 80.87 | 7.9 | 310.27 |
| 9 | 75.61 | 164.04 | 227.48 | 313.97 | 77.56 | 8.49 | 329 |
| 10 | 83.1 | 195.28 | 203.74 | 340.33 | 58.69 | 8.16 | 359.71 |
| 11 | 77.49 | 164.84 | 228.57 | 325.5 | 67.91 | 7.26 | 335 |
| 12 | 84.53 | 171.25 | 229.19 | 308.69 | 91.75 | 11.21 | 439.88 |
| 13 | 81.7 | 171.87 | 225.74 | 112.7 | 284.92 | 8.84 | 312.56 |
| 14_ArH | 84.57 | 173.92 | 226.57 | 338.74 | 61.74 | 11.91 | 383.06 |
| 14_RH | 95.81 | 173.92 | 237.81 | 338.74 | 72.99 | 11.91 | 383.06 |
| BHA | 82.929 | 132.667 | 266.122 | 309.53 | 88.519 | 3.156 | 172.023 |
| Comp. | BDE | IP | PDE | PA | ETE |
|---|---|---|---|---|---|
| 5 | 78.8146275 | 169.60321 | 225.12943 | 300.0272025 | 94.7054375 |
| 9 | 78.2611725 | 135.4063425 | 258.7728425 | 283.7410675 | 110.4381175 |
| 11 | 83.79384 | 136.969445 | 262.7424075 | 297.975905 | 101.7359475 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuelizz, H.A.; Taie, H.A.A.; Bakheit, A.H.; Marzouk, M.; Abdellatif, M.M.; Al-Salahi, R. Biological Evaluation of 4-(1H-triazol-1-yl)benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study. Appl. Sci. 2021, 11, 11642. https://doi.org/10.3390/app112411642
Abuelizz HA, Taie HAA, Bakheit AH, Marzouk M, Abdellatif MM, Al-Salahi R. Biological Evaluation of 4-(1H-triazol-1-yl)benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study. Applied Sciences. 2021; 11(24):11642. https://doi.org/10.3390/app112411642
Chicago/Turabian StyleAbuelizz, Hatem A., Hanan A. A. Taie, Ahmed H. Bakheit, Mohamed Marzouk, Mohamed M. Abdellatif, and Rashad Al-Salahi. 2021. "Biological Evaluation of 4-(1H-triazol-1-yl)benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study" Applied Sciences 11, no. 24: 11642. https://doi.org/10.3390/app112411642
APA StyleAbuelizz, H. A., Taie, H. A. A., Bakheit, A. H., Marzouk, M., Abdellatif, M. M., & Al-Salahi, R. (2021). Biological Evaluation of 4-(1H-triazol-1-yl)benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study. Applied Sciences, 11(24), 11642. https://doi.org/10.3390/app112411642