
Causative Mechanisms of Childhood and Adolescent Obesity Leading to Adult Cardiometabolic Disease: A Literature Review

 , ,
, ,
Abstract
Featured Application
Abstract
1. Introduction
2. Defining Obesity
3. Epidemiology
4. The Anatomy of Obesity
4.1. Surface Disposition of Somatic Adipose Tissue
4.2. Depth of Adipose Tissue
4.3. Local Effects of Adipose Surplus
4.4. Central Obesity and Metabolically Healthy Obesity
4.5. Histological Aspects
5. Obesity Assessment
5.1. Inferential Methods
5.1.1. Anthropometric Parameters
5.1.2. Skinfold Thickness Measurement
5.2. Methods of Determining Body Composition
5.3. Imaging
6. Determinant Factors of Obesity
6.1. Neurohormonal Regulation of Appetite
6.1.1. Hypothalamic Centers
6.1.2. Adipokines
6.1.3. Gastrointestinal Tract Peptides
6.1.4. Other Factors
6.2. Obesity as a Symptom
6.2.1. Genetic Syndromes
6.2.2. Monogenic Causes
6.2.3. Endocrine Disorders
6.2.4. Iatrogenic Obesity
6.3. Genetic Predisposition
6.4. Vulnerable Periods
6.4.1. Pregnancy
6.4.2. New-Born Period and Infancy
6.4.3. Early Childhood, Preschool, and School-Age Periods
6.4.4. Puberty and Adolescence
6.5. Energy Balance
6.5.1. Caloric Intake
6.5.2. Energy Expenditure
6.6. Psychological Aspects
6.7. Social Background
7. Childhood Obesity as an Adult Risk Factor
8. Mechanisms of Obesity-Related Cardiometabolic Disease
9. Obesity Biomarkers and Risk Assessment
9.1. Genetic and Epigenetic Biomarkers
9.2. Inflammatory Markers
9.3. Serological Markers
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Org. Obesity and Overweight. 9 June 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 11 November 2021).
- Kelly, T.; Yang, W.; Chen, C.S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.A.; Trogdon, J.G.; Cohen, J.W.; Dietz, W. Annual medical spending attributable to obesity: Payer-and service-specific estimates. Health Aff. 2009, 28, w822–w831. [Google Scholar] [CrossRef]
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212. [Google Scholar] [CrossRef]
- Ortega, F.B.; Blair, S.N.; Lavie, C.J. Obesity and cardiovascular disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef]
- Falkner, B. Monitoring and management of hypertension with obesity in adolescents. Integr. Blood Press Control 2017, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, M.M.; Zaepfel, A.; Bjornstad, P.; Nadeau, K.J. Age-related consequences of childhood obesity. Gerontology 2014, 60, 222–228. [Google Scholar] [CrossRef]
- Simmonds, M.; Llewellyn, A.; Owen, C.G.; Woolacott, N. Predicting adult obesity from childhood obesity: A systematic review and meta-analysis. Obes. Rev. 2016, 17, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Wolfe, R.; Stoelwinder, J.U.; de Courten, M.; Stevenson, C.; Walls, H.L.; Peeters, A. The number of years lived with obesity and the risk of all-cause and cause-specific mortality. Int. J. Epidemiol. 2011, 40, 985–996. [Google Scholar] [CrossRef]
- World Health Org. Obesity Overview. Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 29 September 2021).
- World Health Org. Adolescent Health. Available online: https://www.who.int/health-topics/adolescent-health#tab=tab_1 (accessed on 15 November 2021).
- Eneli, I.; Dele Davis, H. Epidemiology of childhood obesity. In Obesity in Childhood & Adolescence; Dele Davis, H., Ed.; Praeger Publishers: Westport, CT, USA, 2008; Volume 1, pp. 3–19. [Google Scholar]
- Ortega, F.B.; Labayen, I.; Ruiz, J.R.; Kurvinen, E.; Loit, H.M.; Harro, J.; Veidebaum, T.; Sjöström, M. Improvements in fitness reduce the risk of becoming overweight across puberty. Med. Sci. Sports Exerc. 2011, 43, 1891–1897. [Google Scholar] [CrossRef]
- CDC. CDC Growth Charts. Available online: https://www.cdc.gov/growthcharts/clinical_charts.htm (accessed on 29 September 2021).
- Noncommunicable Diseases: Childhood Overweight and Obesity. World Health Org. 19 October 2020. Available online: https://www.who.int/news-room/q-a-detail/noncommunicable-diseases-childhood-overweight-and-obesity (accessed on 29 September 2021).
- Kelly, A.S.; Barlow, S.E.; Rao, G.; Inge, T.H.; Hayman, L.L.; Steinberger, J.; Urbina, E.M.; Ewing, L.J.; Daniels, S.R.; American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young, Council on Nutrition, Physical Activity and Metabolism, and Council on Clinical Cardiology. Severe obesity in children and adolescents: Identification, associated health risks, and treatment approaches: A scientific statement from the American Heart Association. Circulation 2013, 128, 1689–1712. [Google Scholar] [CrossRef]
- Skinner, A.C.; Skelton, J.A. Prevalence and trends in obesity and severe obesity among children in the United States, 1999–2012. JAMA Pediatr. 2014, 168, 561–566. [Google Scholar] [CrossRef]
- Wang, Y.; Lim, H. The global childhood obesity epidemic and the association between socio-economic status and childhood obesity. Int. Rev. Psychiatry 2012, 24, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.Á.; De Cossío, T.G.; Pedraza, L.S.; Aburto, T.C.; Sánchez, T.G.; Martorell, R. Childhood and adolescent overweight and obesity in Latin America: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 321–332. [Google Scholar] [CrossRef]
- Mazidi, M.; Banach, M.; Kengne, A.P. Prevalence of childhood and adolescent overweight and obesity in Asian countries: A systematic review and meta-analysis. Arch. Med. Sci. 2018, 14, 1185–1203. [Google Scholar] [CrossRef]
- Klingberg, S.; Draper, C.E.; Micklesfield, L.K.; Benjamin-Neelon, S.E.; van Sluijs, E.M.F. Childhood Obesity Prevention in Africa: A Systematic Review of Intervention Effectiveness and Implementation. Int. J. Environ. Res. Public Health 2019, 16, 1212. [Google Scholar] [CrossRef] [PubMed]
- Australian Institute of Health and Welfare. A Picture of Overweight and Obesity in Australia. Cat. no.PHE 216. 2017; 60p. Available online: https://www.aihw.gov.au/getmedia/172fba28-785e-4a08-ab37-2da3bbae40b8/aihw-phe-216.pdf.aspx?inline=true (accessed on 29 September 2021).
- Garrido-Miguel, M.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Rodríguez-Artalejo, F.; Moreno, L.A.; Ruiz, J.R.; Aherens, W.; Martinez-Vizcaíno, V. Prevalence and Trends of Overweight and Obesity in European Children from 1999 to 2016: A Systematic Review and Meta-analysis. JAMA Pediatr. 2019, 173, e192430. [Google Scholar] [CrossRef]
- Janjic, D. “Obésité de type androïde et obésité de type gynoïde” [Android-type obesity and gynecoid-type obesity]. Praxis 1996, 85, 1578–1583. [Google Scholar] [PubMed]
- Ramirez, M.E.; McMurry, M.P.; Wiebke, G.A.; Felten, K.J.; Ren, K.; Meikle, A.W.; Iverius, P.H. Evidence for sex steroid inhibition of lipoprotein lipase in men: Comparison of abdominal and femoral adipose tissue. Metabolism 1997, 46, 179–185. [Google Scholar] [CrossRef]
- Pedersen, S.B.; Kristensen, K.; Hermann, P.A.; Katzenellenbogen, J.A.; Richelsen, B. Estrogen controls lipolysis by up-regulating alpha2A-adrenergic receptors directly in human adipose tissue through the estrogen receptor alpha. Implications for the female fat distribution. J. Clin. Endocrinol. Metab. 2004, 89, 1869–1878. [Google Scholar] [CrossRef]
- Singh, R.; Artaza, J.N.; Taylor, W.E.; Braga, M.; Yuan, X.; Gonzalez-Cadavid, N.F.; Bhasin, S. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: Nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology 2006, 147, 141–154. [Google Scholar] [CrossRef]
- Lacasa, D.; Le Liepvre, X.; Ferre, P.; Dugail, I. Progesterone stimulates adipocyte determination and differentiation 1/sterol regulatory element-binding protein 1c gene expression. potential mechanism for the lipogenic effect of progesterone in adipose tissue. J. Biol. Chem. 2001, 276, 11512–11516. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, V.; Sbraccia, P. Obesity phenotypes: Depot-differences in adipose tissue and their clinical implications. Eat Weight Disord. 2018, 23, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, M. Obesity risk: Importance of the waist–to–height ratio. Nurs. Stand. 2009, 23, 49–54. [Google Scholar] [CrossRef]
- Anderson, P.J.; Chan, J.C.; Chan, Y.L.; Tomlinson, B.; Young, R.P.; Lee, Z.S.; Lee, K.K.C.; Metreweli, C.; Cockram, C.S.; Critchley, J.A.J.H. Visceral fat and cardiovascular risk factors in Chinese NIDDM patients. Diabetes Care 1997, 20, 1854–1858. [Google Scholar] [CrossRef]
- Després, J.P. Body fat distribution and risk of cardiovascular disease an update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef]
- Sironi, A.M.; Petz, R.; De Marchi, D.; Buzzigoli, E.; Ciociaro, D.; Positano, V.; Lombardi, M.; Ferrannini, E.; Gastaldelli, A. Impact of increased visceral and cardiac fat on cardiometabolic risk and disease. Diabet. Med. 2012, 29, 622–627. [Google Scholar] [CrossRef] [PubMed]
- St St-Pierre, J.; Lemieux, I.; Vohl, M.C.; Perron, P.; Tremblay, G.; Després, J.P.; Gaudet, D. Contribution of abdominal obesity and hypertriglyceridemia to impaired fasting glucose and coronary artery disease. Am. J. Cardiol. 2007, 99, 369–373. [Google Scholar] [CrossRef]
- Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular disease: Risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol. 2009, 53, 1925–1932. [Google Scholar] [CrossRef]
- Klöting, N.; Blüher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef]
- Koliaki, C.; Liatis, S.; Kokkinos, A. Obesity and cardiovascular disease: Revisiting an old relationship. Metabolism 2019, 92, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Sugerman, H.J. Effects of increased intra-abdominal pressure in severe obesity. Surg. Clin. N. Am. 2001, 81, 1063–1075. [Google Scholar] [CrossRef]
- Apovian, C.M.; Bigornia, S.; Mott, M.; Meyers, M.R.; Ulloor, J.; Gagua, M.; McDonnell, M.; Hess, D.; Joseph, L.; Gokce, N. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1654–1659. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef]
- Capeau, J. Insulin resistance and steatosis in humans. Diabetes Metab. 2008, 34, 649–657. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Localization of fat depots and cardiovascular risk. Lipids Health Dis. 2018, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Cavallero, S.; Patterson, M.; Shen, H.; Xu, J.; Kumar, S.R.; Sucov, H.M. Adipogenesis and epicardial adipose tissue: A novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2070–2075. [Google Scholar] [CrossRef]
- Manzella, D.; Barbieri, M.; Rizzo, M.R.; Ragno, E.; Passariello, N.; Gambardella, A.; Marfella, R.; Giugliano, D.; Paolisso, G. Role of free fatty acids on cardiac autonomic nervous system in noninsulin-dependent diabetic patients: Effects of metabolic control. J. Clin. Endocrinol. Metab. 2001, 86, 2769–2774. [Google Scholar] [CrossRef] [PubMed]
- Mahabadi, A.A.; Massaro, J.M.; Rosito, G.A.; Levy, D.; Murabito, J.M.; Wolf, P.A.; O’Donnell, C.J.; Fox, C.S.; Hoffmann, U. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: The Framingham heart study. Eur. Heart J. 2009, 30, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Corradi, D.; Maestri, R.; Callegari, S.; Pastori, P.; Goldoni, M.; Luong, T.V.; Bordi, C. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc. Pathol. 2004, 13, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, S.; Sade, L.E.; Yildirir, A.; Bal, U.; Ozbicer, S.; Ozgul, A.S.; Bozbas, H.; Aydinalp, A.; Muderrisoglu, H. Epicardial adipose tissue thickness by echocardiography is a marker for the presence and severity of coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 211–217. [Google Scholar] [CrossRef]
- Wang, C.P.; Hsu, H.L.; Hung, W.C.; Yu, T.H.; Chen, Y.H.; Chiu, C.A.; Lu, L.F.; Chung, F.M.; Shin, S.J.; Lee, Y.J. Increased epicardial adipose tissue (EAT) volume in type 2 diabetes mellitus and association with metabolic syndrome and severity of coronary atherosclerosis. Clin. Endocrinol. 2009, 70, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Butcovan, D.; Mocanu, V.; Timofte, D.V.; Costan, V.V.; Danila, R.; Veselin, A.P.; Ciuntu, B.M.; Haliga, R.E.; Sascau, R.A.; Ghiga, G.; et al. Macrophage Accumulation and Angiogenesis in Epicardial Adipose Tissue in Cardiac Patients with or without Chronic Heart Failure. Appl. Sci. 2020, 10, 5871. [Google Scholar] [CrossRef]
- Rittig, K.; Staib, K.; Machann, J.; Böttcher, M.; Peter, A.; Schick, F.; Claussen, C.; Stefan, N.; Fritsche, A.; Häring, H.U.; et al. Perivascular fatty tissue at the brachial artery is linked to insulin resistance but not to local endothelial dysfunction. Diabetologia 2008, 51, 2093–2099. [Google Scholar] [CrossRef]
- Ashwell, M.; Gunn, P.; Gibson, S. Waist-to-height ratio is a better screening tool than waist circumference and BMI for adult cardiometabolic risk factors: Systematic review and meta-analysis. Obes. Rev. 2012, 13, 275–286. [Google Scholar] [CrossRef]
- Lee, C.M.; Huxley, R.R.; Wildman, R.P.; Woodward, M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than BMI: A meta-analysis. J. Clin. Epidemiol. 2008, 61, 646–653. [Google Scholar] [CrossRef]
- Xue, R.; Li, Q.; Geng, Y.; Wang, H.; Wang, F.; Zhang, S. Abdominal obesity and risk of CVD: A dose-response meta-analysis of thirty-one prospective studies. Br. J. Nutr. 2021, 126, 1420–1430. [Google Scholar] [CrossRef]
- Bosomworth, N.J. Normal-weight central obesity: Unique hazard of the toxic waist. Can. Fam. Physician 2019, 65, 399–408. [Google Scholar] [PubMed]
- Grigorakis, D.A.; Georgoulis, M.; Psarra, G.; Tambalis, K.D.; Panagiotakos, D.B.; Sidossis, L.S. Prevalence and lifestyle determinants of central obesity in children. Eur. J. Nutr. 2016, 55, 1923–1931. [Google Scholar] [CrossRef]
- Canoy, D.; Boekholdt, S.M.; Wareham, N.; Luben, R.; Welch, A.; Bingham, S.; Buchan, I.; Day, N.; Khaw, K.T. Body fat distribution and risk of coronary heart disease in men and women in the European Prospective Investigation Into Cancer and Nutrition in Norfolk cohort: A population-based prospective study. Circulation 2007, 116, 2933–2943. [Google Scholar] [CrossRef]
- Despres, J.P.; Lemieux, I.; Bergeron, J.; Pibarot, P.; Mathieu, P.; Larose, E.; Rodes-Cabau, J.; Bertrand, O.F.; Poirier, P. Abdominal obesity and the metabolic syndrome: Contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Krekoukia, M.; Nassis, G.P.; Psarra, G.; Skenderi, K.; Chrousos, G.P.; Sidossis, L.S. Elevated total and central adiposity and low physical activity are associated with insulin resistance in children. Metab. Clin. Exp. 2007, 56, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Olza, J.; Aguilera, C.M.; Gil-Campos, M.; Leis, R.; Bueno, G.; Valle, M.; Canete, R.; Tojo, R.; Moreno, L.A.; Gil, A. Waist-to-height ratio, inflammation and CVD risk in obese children. Public Health Nutr. 2014, 17, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Manios, Y.; Moschonis, G.; Kourlaba, G.; Bouloubasi, Z.; Grammatikaki, E.; Spyridaki, A.; Hatzis, C.; Kafatos, A.; Fragiadakis, G.A. Prevalence and independent predictors of insulin resistance in children from Crete, Greece: The children study. Diabet. Med. 2008, 25, 65–72. [Google Scholar] [CrossRef]
- Kollias, A.; Psilopatis, I.; Karagiaouri, E.; Glaraki, M.; Grammatikos, E.; Grammatikos, E.E.; Garoufi, A.; Stergiou, G.S. Adiposity, blood pressure, and carotid intima-media thickness in greek adolescents. Obesity 2013, 21, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Shirasawa, T.; Nishimura, R.; Yoshimoto, T.; Minoura, A.; Oikawa, K.; Miki, A.; Hoshino, H.; Kokaze, A. Changes in overweight/obesity and central obesity status from preadolescence to adolescence: A longitudinal study among schoolchildren in Japan. BMC Public Health 2020, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- Hassapidou, M.; Tzotzas, T.; Makri, E.; Pagkalos, I.; Kaklamanos, I.; Kapantais, E.; Abrahamian, A.; Polymeris, A.; Tziomalos, K. Prevalence and geographic variation of abdominal obesity in 7- and 9-year-old children in Greece; World Health Organization childhood obesity surveillance initiative 2010. BMC Public Health 2017, 17, 126. [Google Scholar] [CrossRef]
- Leitao, R.; Rodrigues, L.P.; Neves, L.; Carvalho, G.S. Changes in adiposity status from childhood to adolescence: A 6-year longitudinal study in Portuguese boys and girls. Ann. Hum. Biol. 2011, 38, 520–528. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chrzanowska, M.; Suder, A.; Kruszelnicki, P. Tracking and risk of abdominal obesity in the adolescence period in children aged 7–15. The Cracow longitudinal growth study. Am. J. Hum. Biol. 2012, 24, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.P.; Li, Z.X.; Yang, L.; Zhao, M.; Xi, B. Effect of abdominal obesity in childhood on abdominal obesity in adulthood. Zhonghua Liu Xing Bing Xue Za Zhi 2020, 41, 385–388. (In Chinese) [Google Scholar] [CrossRef]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129(10), 4022–4031. [Google Scholar] [CrossRef] [PubMed]
- Caleyachetty, R.; Thomas, G.N.; Toulis, K.A.; Mohammed, N.; Gokhale, K.M.; Balachandran, K.; Nirantharakumar, K. Metabolically Healthy Obese and Incident Cardiovascular Disease Events Among 3. 5 Million Men and Women. J. Am. Coll. Cardiol. 2017, 70, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, O. The role of adipose tissue as an endocrine gland. Curr. Diab. Rep. 2005, 5, 317–319. [Google Scholar] [CrossRef]
- Aronne, L.J. Classification of obesity and assessment of obesity-related health risks. Obes. Res. 2002, 10, 105S–115S. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.; Blonde, L.; Rosenson, R. Adiposopathy: How do diet, exercise and weight loss drug therapies improve metabolic disease in overweight patients? Expert Rev. Cardiovasc. Ther. 2006, 4, 871–895. [Google Scholar] [CrossRef]
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic; World Health Organization: Geneva, Switzerland, 2000. [Google Scholar]
- Holley, T.J.; Collins, C.E.; Morgan, P.J.; Callister, R.; Hutchesson, M.J. Weight expectations, motivations for weight change and perceived factors influencing weight management in young Australian women: A crosssectional study. Public Health Nutr. 2016, 19, 275–286. [Google Scholar] [CrossRef]
- Soheilipour, F.; Hatami, M.; Salehiniya, H.; Alaei, M. Indicators of Obesity and Cardio-Metabolic Risks: Important Consideration in Adults and Children. Curr. Diabetes Rev. 2021. e-pub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Region WWP. _The Asia-Pacific Perspective: Redefining Obesity and Its Treatment_; International Association for the Study of Obesity, International Obesity Task Force; WHO Western Pacific Region: Geneva, Switzerland, 2000.
- Horan, M.; Gibney, E.; Molloy, E.; McAuliffe, F. Methodologies to assess paediatric adiposity. Ir. J. Med. Sci. 2015, 184, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Xue, B.; Wang, Y. Associations between general and central obesity and hypertension among children: The Childhood Obesity Study in China Mega-Cities. Sci. Rep. 2017, 7, 16895. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Lu, Q.; Wang, R.; Yin, F. Using height-corrected definition of metabolic syndrome in children and adolescents. J. Pediatr. Endocrinol. Metab. 2019, 32, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Emdin, C.A.; Khera, A.V.; Natarajan, P.; Klarin, D.; Zekavat, S.M.; Hsiao, A.J.; Kathiresan, S. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA 2017, 317, 626–634. [Google Scholar] [CrossRef]
- Krakauer, N.Y.; Krakauer, J.C. An Anthropometric Risk Index Based on Combining Height, Weight, Waist, and Hip Measurements. J. Obes. 2016, 2016, 8094275. [Google Scholar] [CrossRef] [PubMed]
- Mameli, C.; Krakauer, N.Y.; Krakauer, J.C.; Bosetti, A.; Ferrari, C.M.; Moiana, N.; Schneider, L.; Borsani, B.; Genoni, T.; Zuccotti, G. The association between a body shape index and cardiovascular risk in overweight and obese children and adolescents. PLoS ONE 2018, 13, e0190426. [Google Scholar] [CrossRef]
- Arias Téllez, M.J.; Martinez-Tellez, B.; Soto, J.; Sánchez-Delgado, G. Validez del perímetro del cuello como marcador de adiposidad en niños, adolescentes y adultos: Una revisión sistemática [Validity of neck circumference as a marker of adiposity in children and adolescents, and in adults: A systematic review]. Nutr. Hosp. 2018, 35, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Akın, O.; Arslan, M.; Haymana, C.; Karabulut, E.; Hacihamdioglu, B.; Yavuz, S.T. Association of neck circumference and pulmonary function in children. Ann. Allergy Asthma Immunol. 2017, 119, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Floras, J.S. Sleep apnea and cardiovascular disease. Circ. Res. 2018, 122, 1741–1764. [Google Scholar] [CrossRef] [PubMed]
- Oppliger, R.A.; Clark, R.R.; Kuta, J.M. Efficacy of skinfold training clinics: A comparison between clinic trained and experienced testers. Res. Q. Exerc. Sport 1992, 63, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Durnin, J.; Womersley, J. Body fat assessed from total body density and its estimation from skinfold thickness: Measurements on 481 men and women aged from 16 to 72 years. Br. J. Nutr. 1974, 32, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.S.; Pollock, M.L.; Ward, A. Generalized equations for predicting body density of women. Med. Sci. Sports Exerc. 1979, 12, 175–181. [Google Scholar] [CrossRef]
- Jackson, A.S.; Pollock, M.L. Generalized equations for predicting body density of men. Br. J. Nutr. 1978, 40, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.J.; Czerwinski, S.A.; Siervogel, R.M. Development and validation of skinfold-thickness prediction equations with a 4-compartment model. Am. J. Clin. Nutr. 2003, 77, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.; Wilson, J.; Durnin, J. Determination of body composition from skinfold thickness: A validation study. Arch. Dis. Child 1995, 73, 305–310. [Google Scholar] [CrossRef]
- Brook, C. Determination of body composition of children from skinfold measurements. Arch. Dis. Child 1971, 46, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, P.; Pieters, J.J.; Hautvast, J.G. The assessment of the body fat percentage by skinfold thickness measurements in childhood and young adolescence. Br. J. Nutr. 1990, 63, 293–303. [Google Scholar] [CrossRef]
- Durnin, J.; Rahaman, M. The assessment of the amount of fat in the human body from measurements of skinfold thickness. Br. J. Nutr. 1967, 21, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, M.H.; Lohman, T.; Boileau, R.; Horswill, C.; Stillman, R.; Van Loan, M.; Bemben, D. Skinfold equations for estimation of body fatness in children and youth. Hum. Biol. 1988, 60, 709–723. [Google Scholar] [PubMed]
- Toombs, R.J.; Ducher, G.; Shepherd, J.A.; Souza, M.J. The impact of recent technological advances on the trueness and precision of DXA to assess body composition. Obesity 2012, 20, 30–39. [Google Scholar] [CrossRef]
- Wells, J.C.; Haroun, D.; Williams, J.E.; Wilson, C.; Darch, T.; Viner, R.M.; Eaton, S.; Fewtrell, M.S. Evaluation of DXA against the four-component model of body composition in obese children and adolescents aged 5–21 years. Int. J. Obes. 2010, 34, 649–655. [Google Scholar] [CrossRef][Green Version]
- Ward, L.C.; Poston, L.; Godfrey, K.M.; Koletzko, B. Assessing early growth and adiposity: Report from an EarlyNutrition Academy Workshop. Ann. Nutr. Metab. 2013, 63, 120–130. [Google Scholar] [CrossRef]
- Harrington, T.; Thomas, E.; Modi, N.; Frost, G.; Coutts, G.; Bell, J. Fast and reproducible method for the direct quantitation of adipose tissue in newborn infants. Lipids 2002, 37, 95–100. [Google Scholar] [CrossRef]
- Lukaski, H.C.; Johnson, P.E.; Bolonchuk, W.; Lykken, G. Assessment of fat-free mass using bioelectrical impedance measurements of the human body. Am. J. Clin. Nutr. 1985, 41, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Kyle, U.G.; Bosaeus, I.; De Lorenzo, A.D.; Deurenberg, P.; Elia, M.; Go’mez, J.M.; Heitmann, B.L.; Kent-Smith, L.; Melchior, J.-C.; Pirlich, M. Bioelectrical impedance analysis—Part I: Review of principles and methods. Clin. Nutr. 2004, 23, 1226–1243. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.; Walker, K.; O’Dea, K. The influence of a breakfast meal on the assessment of body composition using bioelectrical impedance. Eur. J. Clin. Nutr. 1998, 52, 94–97. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.; Timmers, T.; Weijs, P.J.; Gemke, R.J. Validation of total body water analysis by bioelectrical impedance analysis with deuterium dilution in (pre) school children. e-SPEN: Eur. e-J. Clin. Nutr. Metab. 2011, 6, e223–e226. [Google Scholar] [CrossRef]
- Shafer, K.J.; Siders, W.A.; Johnson, L.K.; Lukaski, H.C. Validity of segmental multiple-frequency bioelectrical impedance analysis to estimate body composition of adults across a range of body mass indexes. Nutrition 2009, 25, 25–32. [Google Scholar] [CrossRef]
- Lukaski, H.C. Methods for the assessment of human body composition: Traditional and new. Am. J. Clin. Nutr. 1987, 46, 537–556. [Google Scholar] [CrossRef]
- Claros, G.; Hull, H.R.; Fields, D.A. Comparison of air displacement plethysmography to hydrostatic weighing for estimating total body density in children. BMC Pediatr. 2005, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Demerath, E.; Guo, S.; Chumlea, W.; Towne, B.; Roche, A.; Siervogel, R. Comparison of percent body fat estimates using air displacement plethysmography and hydrodensitometry in adults and children. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 389–397. [Google Scholar] [CrossRef]
- Holmes, J.C.; Gibson, A.L.; Cremades, J.G.; Mier, C.M. Bodydensity measurement in children: The BOD POD versus Hydrodensitometry. Int. J. Sport Nutr. Exerc. Metab. 2011, 21, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Caprio, S.; Hyman, L.D.; McCarthy, S.; Lange, R.; Bronson, M.; Tamborlane, W.V. Fat distribution and cardiovascular risk factors in obese adolescent girls: Importance of the intraabdominal fat depot. Am. J. Clin. Nutr. 1996, 64, 12–17. [Google Scholar] [CrossRef]
- Fields, D.A.; Goran, M.I.; McCrory, M.A. Body-composition assessment via air-displacement plethysmography in adults and children: A review. Am. J. Clin. Nutr. 2002, 75, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.P.; Hourihane, J.O.B.; Kenny, L.C.; Irvine, A.D.; Kiely, M.; Murray, D.M. Gender-and gestational age-specific body fat percentage at birth. Pediatrics 2011, 128, e645–e651. [Google Scholar] [CrossRef] [PubMed]
- Fields, D.A.; Allison, D.B. Air-displacement plethysmography pediatric option in 2–6 years old using the four-compartment model as a criterion method. Obesity 2012, 20, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Gately, P.; Radley, D.; Cooke, C.; Carroll, S.; Oldroyd, B.; Truscott, J.; Coward, W.; Wright, A. Comparison of body composition methods in overweight and obese children. J. Appl. Physiol. 2003, 95, 2039–2046. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wells, J.C.; Williams, J.E.; Chomtho, S.; Darch, T.; Grijalva-Eternod, C.; Kennedy, K.; Haroun, D.; Wilson, C.; Cole, T.J.; Fewtrell, M.S. Body-composition reference data for simple and reference techniques and a 4-component model: A new UK reference child. Am. J. Clin. Nutr. 2012, 96, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.R.; Tobkin, S.E.; Costa, P.B.; Smalls, M.; Mieding, W.K.; O’Kroy, J.A.; Zoeller, R.F.; Stout, J.R. Validity of the BOD POD for assessing body composition in athletic high school boys. J. Strength Cond. Res. 2008, 22, 263–268. [Google Scholar] [CrossRef]
- Wells, J.C.; Haroun, D.; Williams, J.E.; Darch, T.; Eaton, S.; Viner, R.; Fewtrell, M. Evaluation of lean tissue density for use in air displacement plethysmography in obese children and adolescents. Eur. J. Clin. Nutr. 2011, 65, 1094–1101. [Google Scholar] [CrossRef][Green Version]
- Fields, D.A.; Goran, M.I. Body composition techniques and the four-compartment model in children. J. Appl. Physiol. 2000, 89, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Bila, W.C.; Freitas, A.E.; Galdino, A.S.; Ferriolli, E.; Pfrimer, K.; Lamounier, J.A. Deuterium oxide dilution and body composition in overweight and obese schoolchildren aged 6–9 years. J. Pediatr. 2016, 92, 46–52. [Google Scholar] [CrossRef][Green Version]
- Koletzko, B.; Demmelmai, H.; Hartl, W.; Kindermann, A.; Koletzko, S.; Sauerwald, T.; Szitanyi, P. The use of stable isotope techniques for nutritional and metabolic research in paediatrics. Early Hum. Dev. 1998, 53 (Suppl. S1), S77–S97. [Google Scholar] [CrossRef]
- De Lucia Rolfe, E.; Modi, N.; Uthaya, S.; Hughes, I.A.; Dunger, D.B.; Acerini, C.; Stolk, R.P.; Ong, K.K. Ultrasound estimates of visceral and subcutaneous-abdominal adipose tissues in infancy. J. Obes. 2013, 2013, 951954. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.R. Ultrasound as a tool to assess body fat. J. Obes. 2013, 2013, 280713. [Google Scholar] [CrossRef] [PubMed]
- Liem, E.; Rolfe, E.D.L.; L’abee, C.; Sauer, P.; Ong, K.; Stolk, R. Measuring abdominal adiposity in 6 to 7-year-old children. Eur. J. Clin. Nutr. 2009, 63, 835–841. [Google Scholar] [CrossRef]
- Koot, B.; Westerhout, R.; Bohte, A.; Vinke, S.; Pels Rijcken, T.; Nederveen, A.; Caan, M.; Baan-Slootweg, O.; Merkus, M.; Stoker, J. Ultrasonography is not more reliable than anthropometry for assessing visceral fat in obese children. Pediatr. Obes 2013, 9, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Mook-Kanamori, D.O.; Holzhauer, S.; Hollestein, L.M.; Durmus, B.; Manniesing, R.; Koek, M.; Boehm, G.; Van der Beek, E.M.; Hofman, A.; Witteman, J.C. Abdominal fat in children measured by ultrasound and computed tomography. Ultrasound Med. Biol. 2009, 35, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Zemel, B.S. Quantitative computed tomography and computed tomography in children. Curr. Osteoporos Rep. 2011, 9, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.K.; Johnson, M.S.; Figueroa-Colon, R.; Dwyer, J.H.; Goran, M.I. Growth of visceral fat, subcutaneous abdominal fat, and total body fat in children. Obes. Res. 2001, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Punyanitya, M.; Wang, Z.; Gallagher, D.; St-Onge, M.-P.; Albu, J.; Heymsfield, S.B.; Heshka, S. Visceral adipose tissue: Relations between single-slice areas and total volume. Am. J. Clin. Nutr. 2004, 80, 271–278. [Google Scholar] [CrossRef]
- Shen, W.; Punyanitya, M.; Wang, Z.; Gallagher, D.; Onge, M.-P.S.; Albu, J.; Heymsfield, S.B.; Heshka, S. Total body skeletal muscle and adipose tissue volumes: Estimation from a single abdominal crosssectional image. J. Appl. Physiol. 2004, 97, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liu, H.; Punyanitya, M.; Chen, J.; Heymsfield, S.B. Pediatric obesity phenotyping by magnetic resonance methods. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 595. [Google Scholar]
- Shen, W.; Chen, J.; Gantz, M.; Velasquez, G.; Punyanitya, M.; Heymsfield, S.B. A single MRI slice does not accurately predict visceral and subcutaneous adipose tissue changes during weight loss. Obesity 2012, 20, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Uthaya, S.; Bell, J.; Modi, N. Adipose tissue magnetic resonance imaging in the newborn. Horm. Res. Paediatr. 2004, 62 (Suppl. S3), 143–148. [Google Scholar] [CrossRef]
- Gale, C.; Jeffries, S.; Logan, K.M.; Chappell, K.E.; Uthaya, S.N.; Modi, N. Avoiding sedation in research MRI and spectroscopy in infants: Our approach, success rate and prevalence of incidental findings. Arch. Dis. Child Fetal Neonatal Ed. 2013, 98, F267–F268. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, C.L.; Rohling, K.W.; Piel, J.E.; Rossi, C.J.; Giaquinto, R.O.; Watkins, R.D.; Vigneron, D.B.; Barkovich, A.J.; Newton, N. Magnetic resonance imaging compatible neonate incubator. Concepts Magn. Reson. 2002, 15, 117–128. [Google Scholar] [CrossRef]
- Samara, A.; Ventura, E.; Alfadda, A.; Goran, M. Use of MRI and CT for fat imaging in children and youth: What have we learned about obesity, fat distribution and metabolic disease risk? Obes. Rev. 2012, 13, 723–732. [Google Scholar] [CrossRef]
- Takatalo, J.; Karppinen, J.; Taimela, S.; Niinimäki, J.; Laitinen, J.; Sequeiros, R.B.; Samartzis, D.; Korpelainen, R.; Näyhä, S.; Remes, J.; et al. Association of abdominal obesity with lumbar disc degeneration—a magnetic resonance imaging study. PLoS ONE 2013, 8, e56244. [Google Scholar] [CrossRef]
- Brown, R.E.; Kuk, J.L.; Lee, S. Measurement site influences abdominal subcutaneous and visceral adipose tissue in obese adolescents before and after exercise. Pediatric. Obes. 2015, 10, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Eloi, J.C.; Epifanio, M.; de Gonçalves, M.M.; Pellicioli, A.; Vieira, P.F.; Dias, H.B.; Bruscato, N.; Soder, R.B.; Santana, J.C.; Mouzaki, M.; et al. Quantification of Abdominal Fat in Obese and Healthy Adolescents Using 3 Tesla Magnetic Resonance Imaging and Free Software for Image Analysis. PLoS ONE 2017, 12, e0167625. [Google Scholar] [CrossRef]
- Binkley, C.M.; Jing, L.; Suever, J.D.; Umasankar, N.; Wehner, G.J.; Hamlet, S.M.; Powell, D.; Radulescu, A.; Epstein, F.H.; Fornwalt, B.K. Children with obesity have cardiac remodeling and dysfunction: A cine DENSE magnetic resonance imaging study. J. Cardiovasc. Magn. Reson. 2015, 17, Q57. [Google Scholar] [CrossRef]
- Orsso, C.E.; Colin-Ramirez, E.; Field, C.J.; Madsen, K.L.; Prado, C.M.; Haqq, A.M. Adipose Tissue Development and Expansion from the Womb to Adolescence: An Overview. Nutrients 2020, 12, 2735. [Google Scholar] [CrossRef]
- Haylett, W.L.; Ferris, W.F. Adipocyte–progenitor cell communication that influences adipogenesis. Cell Mol. Life Sci. 2020, 77, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue infammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Caprio, S.; Santoro, N.; Weiss, R. Childhood obesity and the associated rise in cardiometabolic complications. Nat. Metab. 2020, 2, 223–232. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Sethi, J.K.; Vidal-Puig, A.J. Thematic review series: Adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007, 48, 1253–1262. [Google Scholar] [CrossRef]
- Toemen, L.; Santos, S.; Roest, A.A.; Jelic, G.; van der Lugt, A.; Felix, J.F.; Helbing, W.A.; Gaillard, R.; Jaddoe, V.W.V. Body Fat Distribution, Overweight, and Cardiac Structures in School-Age Children: A Population-Based Cardiac Magnetic Resonance Imaging Study. J. Am. Heart Assoc. 2020, 9, e014933. [Google Scholar] [CrossRef]
- Dencker, M.; Danielson, A.; Karlsson, M.K.; Wollmer, P.; Andersen, L.B.; Thorsson, O. Total body fat, abdominal fat, body fat distribution and surrogate markers for health related to adipocyte fatty acid-binding protein (FABP4) in children. J. Pediatr. Endocrinol. Metab. JPEM 2017, 30, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Farias, G.; Netto, B.; Bettini, S.C.; Dâmaso, A.R.; de Freitas, A. Neuroendocrine regulation of energy balance: Implications on the development and surgical treatment of obesity. Nutr. Health 2017, 23, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Nakazato, M. Mechanistic relationship between the vagal afferent pathway, central nervous system and peripheral organs in appetite regulation. J. Diabetes Investig. 2016, 7, 812–818. [Google Scholar] [CrossRef]
- Farooqi, S.I. Genetic, molecular and physiological mechanisms involved in human obesity: Society for Endocrinology Medal Lecture 2012. Clin. Endocrinol. 2015, 82, 23–28. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Lieb, W.; Sullivan, L.M.; Harris, T.B.; Roubenoff, R.; Benjamin, E.J.; Levy, D.; Fox, C.S.; Wang, T.J.; Wilson, P.W.; Kannel, W.B.; et al. Plasma leptin levels and incidence of heart failure, cardiovascular disease, and total mortality in elderly individuals. Diabetes Care 2009, 32, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Re. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef]
- Shah, M.; Vella, A. Effects of GLP-1 on appetite and weight. Rev. Endocr. Metab. Disord. 2014, 15, 181–187. [Google Scholar] [CrossRef]
- Kokot, F.; Ficek, R. Effects of neuropeptide Y on appetite. Min. Electrol. Metab. 1999, 25, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Channa, N.J.; Bloom, S.R. The Gut Hormones in Appetite Regulation. J. Obes. 2011, 2011, 528401. [Google Scholar] [CrossRef]
- Cummings, D.E.; Shannon, M.H. Roles for Ghrelin in the Regulation of Appetite and Body Weight. Arch. Surg. 2003, 138, 389–396. [Google Scholar] [CrossRef]
- Kirkham, T.C. Endocannabinoids in the regulation of appetite and body weight. Behav. Pharmacol. 2005, 16, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, C.; Heni, M.; Thamer, C.; Herzberg-Schäfer, S.A.; Häring, H.U.; Fritsche, A. Acute, short-term hyperinsulinemia increases olfactory threshold in healthy subjects. Int. J. Obes. 2011, 35, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Lobstein, T.; Baur, L.; Uauy, R. Obesity in children and young people: A crisis in public health. Obes. Rev. 2004, 5 (Suppl. S1), 4–104. [Google Scholar] [CrossRef]
- Gluckman, P.; Nishtar, S.; Armstrong, T. Ending childhood obesity: A multidimensional challenge. Lancet 2015, 385, 1048–1050. [Google Scholar] [CrossRef]
- Cecil, J.E.; Tavendale, R.; Watt, P.; Hetherington, M.M.; Palmer, C.N. An obesity-associated FTO gene variant and increased energy intake in children. N. Engl. J. Med. 2008, 359, 2558–2566. [Google Scholar] [CrossRef]
- Qi, Q.; Chu, A.Y.; Kang, J.H.; Huang, J.; Rose, L.M.; Jensen, M.K.; Liang, L.; Curhan, G.C.; Pasquale, L.R.; Wiggs, J.L.; et al. Fried food consumption, genetic risk, and body mass index: Gene-diet interaction analysis in three US cohort studies. BMJ. 2014, 348, g1610. [Google Scholar] [CrossRef]
- Felix, J.F.; Bradfield, J.P.; Monnereau, C.; van der Valk, R.J.; Stergiakouli, E.; Chesi, A.; Gaillard, R.; Feenstra, B.; Thiering, E.; Kreiner-Møller, E.; et al. Genome-wide association analysis identifes three new susceptibility loci for childhood body mass index. Hum. Mol. Genet. 2016, 25, 389–403. [Google Scholar] [CrossRef]
- Bradfeld, J.P.; Taal, H.R.; Timpson, N.J.; Scherag, A.; Lecoeur, C.; Warrington, N.M.; Hypponen, E.; Holst, C.; Valcarcel, B.; Thiering, E.; et al. A genome-wide association meta-analysis identifes new childhood obesity loci. Nat. Genet. 2012, 44, 526–531. [Google Scholar] [CrossRef]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Investig. 2017, 127, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.S.; Creemers, J.W.; Ohagi, S.; Raffin-Sanson, M.L.; Sanders, L.; Montague, C.T.; Hutton, J.C.; O’Rahilly, S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 1997, 16, 303–306. [Google Scholar] [CrossRef]
- Paisey, R.B.; Steeds, R.; Barrett, T.; Williams, D.; Geberhiwot, T.; Gunay-Aygun, M. Alström Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Han, J.C.; Reyes-Capo, D.P.; Liu, C.Y.; Reynolds, J.C.; Turkbey, E.; Turkbey, I.B.; Bryant, J.; Marshall, J.D.; Naggert, J.K.; Gahl, W.A.; et al. Comprehensive endocrine-metabolic evaluation of patients with Alström syndrome compared with BMI-matched controls. J. Clin. Endocrinol. Metab. 2018, 103, 2707–2719. [Google Scholar] [CrossRef]
- Sherafat-Kazemzadeh, R.; Ivey, L.; Kahn, S.R.; Sapp, J.C.; Hicks, M.D.; Kim, R.C.; Krause, A.J.; Shomaker, L.B.; Biesecker, L.G.; Han, J.C.; et al. Hyperphagia among patients with Bardet-Biedl syndrome. Pediatr. Obes. 2013, 8, e64–e67. [Google Scholar] [CrossRef] [PubMed]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.; Sharma, R. Childhood obesity. Indian J. Pediatr. 2013, 80, 309–317. [Google Scholar] [CrossRef]
- Swaab, D.F.; Purba, J.S.; Hofman, M.A. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: A study of five cases. J. Clin. Endocrinol. Metab. 1995, 80, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Mykytyn, K.; Nishimura, D.; Searby, C.; Shastri, M.; Yen, H.J.; Beck, J.S.; Braun, T.; Streb, L.M.; Cornier, A.S.; Cox, G.F.; et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 2002, 31, 435–438. [Google Scholar] [CrossRef]
- Tarhan, E.; Oǧuz, H.; Şafak, M.A.; Samim, E. The Carpenter syndrome phenotype. Int. J. Pediatr. Otorhinolaryngol. 2004, 68, 353–357. [Google Scholar] [CrossRef]
- Lodhia, J.; Rego-Garcia, I.; Koipapi, S.; Sadiq, A.; Msuya, D.; Spaendonk, R.V.; Hamel, B.; Dekker, M. Carpenter syndrome in a patient from Tanzania. Am. J. Med. Genet. Part A 2021, 185A, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Langmann, A.; Lindner, S. Cohen syndrome. Spektrum Augenheilkd 1995, 9, 218–220. [Google Scholar] [CrossRef]
- Rodrigues, J.M.; Fernandes, H.D.; Caruthers, C.; Braddock, S.R.; Knutsen, A.P. Cohen Syndrome: Review of the Literature. Cureus 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Kaya, A.; Orbak, Z.; Ca̧yir, A.; Döneray, H.; Taşdemir, Ş.; Ozanẗurk, A.; Bingöl, F. Combined occurrence of Alström syndrome and bronchiectasis. Pediatrics 2014, 133, e780–e783. [Google Scholar] [CrossRef] [PubMed]
- Kang, S. Adipose Tissue Malfunction Drives Metabolic Dysfunction in Alström Syndrome. Diabetes 2021, 70, 323–325. [Google Scholar] [CrossRef]
- Huvenne, H.; Dubern, B.; Clément, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.T.; Farooqi, I.S.; Moreau, M.; DePaoli, A.M.; Lawrence, E.; O’Rahilly, S.; Trussell, R.A. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: Report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab. 2004, 89, 4821–4826. [Google Scholar] [CrossRef]
- Cummings, D.E.; Schwartz, M.W. Melanocortins and body weight: A tale of two receptors. Nat. Genet. 2000, 26, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Clement, K.; Durand, E.; Hercberg, S.; Guy-Grand, B.; Froguel, P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Investig. 2000, 106, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, S.L. Clinical review 127: Obesity as a neuroendocrine disease: Lessons to be learned from proopiomelanocortin and melanocortin receptor mutations in mice and men. J. Clin. Endocrinol. Metab. 2001, 86, 1442–1446. [Google Scholar] [CrossRef] [PubMed]
- Celi, F.S.; Shuldiner, A.R. The role of peroxisome proliferator-activated receptor gamma in diabetes and obesity. Curr. Diab. Rep. 2002, 2, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Karam, J.; McFarlane, S. Secondary causes of obesity. Therapy 2007, 4, 641–650. [Google Scholar] [CrossRef]
- Stipancić, G. Secondary causes of obesity in children and adolescents. Cent. Eur. J. Paediatr. 2018, 14, 1–11. [Google Scholar] [CrossRef]
- Gurnani, M.; Birken, C.; Hamilton, J. Childhood Obesity. Pediatr. Clin. N. Am. 2015, 62, 821–840. [Google Scholar] [CrossRef]
- Del Fiol, F.S.; Balcão, V.M.; Barberato-Fillho, S.; Lopes, L.C.; Bergamaschi, C.C. Obesity: A New Adverse Effect of Antibiotics? Front. Pharmacol. 2018, 9, 1408. [Google Scholar] [CrossRef]
- Robert, M.; Kliegman, M.D.; Geme, J.S. Nelson Textbook of Pediatrics, 21st ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Ranjani, H.; Pradeepa, R.; Mehreen, T.S.; Anjana, R.M.; Anand, K.; Garg, R.; Mohan, V. Determinants, consequences and prevention of childhood overweight and obesity: An Indian context. Indian J. Endocrinol. Metab. 2014, 18 (Suppl. S1), S17–S25. [Google Scholar] [CrossRef]
- Reilly, J.J.; Armstrong, J.; Dorosty, A.R.; Emmett, P.M.; Ness, A.; Rogers, I.; Steer, C.; Sherriff, A.; Avon Longitudinal Study of Parents and Children Study Team. Early life risk factors for obesity in childhood: Cohort study. BMJ 2005, 330, 1357. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Tremblay, A.; Després, J.P.; Nadeau, A.; Lupien, P.J.; Thériault, G.; Dussault, J.; Moorjani, S.; Pinault, S.; Fournier, G. The response to long-term overfeeding in identical twins. N. Engl. J. Med. 1990, 322, 1477–1482. [Google Scholar] [CrossRef]
- Sørensen, T.I.; Holst, C.; Stunkard, A.J.; Skovgaard, L.T. Correlations of body mass index of adult adoptees and their biological and adoptive relatives. Int. J. Obes. 1992, 16, 227–236. [Google Scholar]
- Freeman, E.; Fletcher, R.; Collins, C.E.; Morgan, P.J.; Burrows, T.; Callister, R. Preventing and treating childhood obesity: Time to target fathers. Int. J. Obes. 2012, 36, 12–15. [Google Scholar] [CrossRef]
- Neel, J.V. The “thrifty genotype” in 1998. Nutr. Rev. 1999, 57 Pt 2, S2–S9. [Google Scholar] [CrossRef]
- Hunter, D.J. Gene-environment interactions in human diseases. Nat. Rev. Genet. 2005, 6, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. Fetal origins of coronary heart disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsen, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Dietz, W.H. Overweight in childhood and adolescence. N. Engl. J. Med. 2004, 350, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Labayen, I.; Ruiz, J.R.; Vicente-Rodríguez, G.; Turck, D.; Rodríguez, G.; Meirhaeghe, A.; Molnár, D.; Sjöström, M.; Castillo, M.J.; Gottrand, F.; et al. Healthy Lifestyle in Europe by Nutrition in Adolescence (HELENA) Study Group. Early life programming of abdominal adiposity in adolescents: The HELENA Study. Diabetes Care 2009, 32, 2120–2122. [Google Scholar] [CrossRef]
- Vidakovic, A.J.; Gishti, O.; Voortman, T.; Felix, J.F.; Williams, M.A.; Hofman, A.; Demmelmair, H.; Koletzko, B.; Tiemeier, H.; Jaddoe, V.W.; et al. Maternal plasma PUFA concentrations during pregnancy and childhood adiposity: The Generation R Study. Am. J. Clin. Nutr. 2016, 103, 1017–1025. [Google Scholar] [CrossRef]
- Hakola, L.; Takkinen, H.M.; Niinistö, S.; Ahonen, S.; Erlund, I.; Rautanen, J.; Veijola, R.; Ilonen, J.; Toppari, J.; Knip, M.; et al. Maternal fatty acid intake during pregnancy and the development of childhood overweight: A birth cohort study. Pediatr. Obes. 2016, 12, S26–S37. [Google Scholar] [CrossRef] [PubMed]
- Gjestland, K.; Bo, K.; Owe, K.M.; Eberhard-Gran, M. Do pregnant women follow exercise guidelines? Prevalence data among 3482 women, and prediction of low-back pain, pelvic girdle pain and depression. Br. J. Sports Med. 2013, 47, 515–520. [Google Scholar] [CrossRef]
- Evenson, K.R.; Savitz, D.A.; Huston, S.L. Leisuretime physical activity among pregnant women in the US. Paediatr. Perinat. Epidemiol. 2004, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Brinton, J.T.; Glueck, D.H.; Shapiro, A.L.; Harrod, C.S.; Lynch, A.M.; Siega-Riz, A.M.; Dabelea, D. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start Study. Am. J. Clin. Nutr. 2015, 101, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Aris, I.M.; Tint, M.T.; Soh, S.E.; Godfrey, K.M.; Yeo, G.S.H.; Kwek, K.; Chan, J.K.Y.; Gluckman, P.D.; Chong, Y.S.; et al. Ethnic differences in effects of maternal pre-pregnancy and pregnancy adiposity on offspring size and adiposity. J. Clin. Endocrinol. Metab. 2015, 100, 3641–3650. [Google Scholar] [CrossRef]
- Castillo, H.; Santos, I.S.; Matijasevich, A. Relationship between maternal pre- pregnancy body mass index, gestational weight gain and childhood fatness at 6–7 years by air displacement plethysmography. Matern. Child Nutr. 2015, 11, 606–617. [Google Scholar] [CrossRef]
- Hivert, M.F.; Rifas-Shiman, S.L.; Gillman, M.W.; Oken, E. Greater early and mid- pregnancy gestational weight gains are associated with excess adiposity in mid-childhood. Obesity 2016, 24, 1546–1553. [Google Scholar] [CrossRef]
- Kral, J.G.; Biron, S.; Simard, S.; Hould, F.S.; Lebel, S.; Marceau, S.; Marceau, P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 2006, 118, e1644–e1649. [Google Scholar] [CrossRef]
- Smith, J.; Cianflone, K.; Biron, S.; Hould, F.S.; Lebel, S.; Marceau, S.; Lescelleur, O.; Biertho, L.; Simard, S.; Kral, J.G.; et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J. Clin. Endocrinol. Metab. 2009, 94, 4275–4283. [Google Scholar] [CrossRef] [PubMed]
- Branum, A.M.; Parker, J.D.; Keim, S.A.; Schempf, A.H. Prepregnancy body mass index and gestational weight gain in relation to child body mass index among siblings. Am. J. Epidemiol. 2011, 174, 1159–1165. [Google Scholar] [CrossRef]
- Larqué, E.; Labayen, I.; Flodmark, C.E.; Lissau, I.; Czernin, S.; Moreno, L.A.; Pietrobelli, A.; Widhalm, K. From conception to infancy—early risk factors for childhood obesity. Nat. Rev. Endocrinol. 2019, 15, 456–478. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Levitan, E.B.; Gillman, M.W. Maternal smoking during pregnancy and child overweight: Systematic review and meta- analysis. Int. J. Obes. 2008, 32, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Kurniawan, N.D.; Yamada, L.; Fleming, W.; Kaminen-Ahola, N.; Ahola, A.; Galloway, G.; Chong, S. Early gestational ethanol exposure in mice: Effects on brain structure, energy metabolism and adiposity in adult offspring. Alcohol 2018, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.Y.; Kim, S.H.; Park, M.J. Air pollution and childhood obesity. Clin. Exp. Pediatr. 2020, 63, 382–388. [Google Scholar] [CrossRef]
- Lupattelli, A.; Spigset, O.; Twigg, M.J.; Zagorodnikova, K.; Mårdby, A.C.; Moretti, M.E.; Drozd, M.; Panchaud, A.; Hämeen-Anttila, K.; Rieutord, A.; et al. Medication use in pregnancy: A cross- sectional, multinational web- based study. BMJ Open 2014, 4, e004365. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Murphy, S.K.; Murtha, A.P.; Schildkraut, J.M.; Soubry, A.; Huang, Z.; Neelon, S.E.; Fuemmeler, B.; Iversen, E.; Wang, F.; et al. Associations between antibiotic exposure during pregnancy, birth weight and aberrant methylation at imprinted genes among offspring. Int. J. Obes. 2013, 37, 907–913. [Google Scholar] [CrossRef]
- Jepsen, P.; Skriver, M.V.; Floyd, A.; Lipworth, L.; Schønheyder, H.C.; Sørensen, H.T. A population- based study of maternal use of amoxicillin and pregnancy outcome in Denmark. Br. J. Clin. Pharmacol. 2003, 55, 216–221. [Google Scholar] [CrossRef]
- Mor, A.; Antonsen, S.; Kahlert, J.; Holsteen, V.; Jørgensen, S.; Holm-Pedersen, J.; Sørensen, H.T.; Pedersen, O.; Ehrenstein, V. Prenatal exposure to systemic antibacterials and overweight and obesity in Danish schoolchildren: A prevalence study. Int. J. Obes. 2015, 39, 1450–1455. [Google Scholar] [CrossRef]
- Logan, K.M.; Gale, C.; Hyde, M.J.; Santhakumaran, S.; Modi, N. Diabetes in pregnancy and infant adiposity: Systematic review and meta- analysis. Arch. Dis. Child Fetal Neonatal Ed. 2017, 102, F65–F72. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Lichtenstein, P.; Langstrom, N. Association of maternal diabetes mellitus in pregnancy with offspring adiposity into early adulthood: Sibling study in a prospective cohort of 280,866 men from 248,293 families. Circulation 2011, 123, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Gaskins, A.J.; Blaine, A.I.; Zhang, C.; Gillman, M.W.; Missmer, S.A.; Field, A.E.; Chavarro, J.E. Association Between Cesarean Birth and Risk of Obesity in Offspring in Childhood, Adolescence, and Early Adulthood. JAMA Pediatr. 2016, 170, e162385. [Google Scholar] [CrossRef] [PubMed]
- Section on Breastfeeding. Breastfeeding and the use of human milk. Pediatrics 2012, 129, e827–e841. [Google Scholar] [CrossRef] [PubMed]
- ESPGHAN Committee on Nutrition; Agostoni, C.; Braegger, C.; Decsi, T.; Kolacek, S.; Koletzko, B.; Michaelsen, K.F.; Mihatsch, W.; Moreno, L.A.; Puntis, J.; et al. Breast-feeding: A commentary by the ESPGHAN Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 112–125. [Google Scholar] [CrossRef]
- World Health Org. Breastfeeding. Available online: https://www.who.int/health-topics/breastfeeding#tab=tab_2 (accessed on 29 September 2021).
- Kries, V.R.; Koletzko, B.; Sauerwald, T.; Mutius, V.E.; Barnert, D.; Grunert, V.; von Voss, H. Breast feeding and obesity: Cross sectional study. BMJ 1999, 319, 147–150. [Google Scholar] [CrossRef]
- Gillman, M.W.; Rifas-Shiman, S.L.; Camargo, C.A., Jr.; Berkey, C.S.; Frazier, A.L.; Rockett, H.R.; Field, A.E.; Colditz, G.A. Risk of overweight among adolescents who were breastfed as infants. JAMA 2001, 285, 2461–2467. [Google Scholar] [CrossRef]
- World Health Organization. Protecting, Promoting and Supporting Breast-Feeding in Facilities Providing Maternity and Newborn Services; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Daniels, S.R.; Arnett, D.K.; Eckel, R.H.; Gidding, S.S.; Hayman, L.L.; Kumanyika, S.; Robinson, T.N.; Scott, B.J.; St Jeor, S.; Williams, C.L. Overweight in children and adolescents: Pathophysiology, consequences, prevention, and treatment. Circulation 2005, 111, 1999–2012. [Google Scholar] [CrossRef]
- Mueller, W.H. The changes with age of the anatomical distribution of fat. Soc. Sci. Med. 1982, 16, 191–196. [Google Scholar] [CrossRef]
- Morrison, J.A.; Sprecher, D.L.; Barton, B.A.; Waclawiw, M.A.; Daniels, S.R. Overweight, fat patterning, and cardiovascular disease risk factors in black and white girls: The National Heart, Lung, and Blood Institute Growth and Health Study. J. Pediatr. 1999, 135, 458–464. [Google Scholar] [CrossRef]
- Morrison, J.A.; Barton, B.A.; Biro, F.M.; Daniels, S.R.; Sprecher, D.L. Overweight, fat patterning, and cardiovascular disease risk factors in black and white boys. J. Pediatr. 1999, 135, 451–457. [Google Scholar] [CrossRef]
- Juliot, L. Modernité et désarrois de l’adolescence [Modernity and turmoil of adolescence]. Soins Psychiatr. 2020, 41, 39–43. [Google Scholar] [CrossRef]
- U.S. Department of Agriculture; U.S. Department of Health and Human Services. Dietary Guidelines for Americans, 2020-2025. 9th Edition. December 2020. Available online: DietaryGuidelines.gov (accessed on 11 November 2021).
- Ross, A.C.; Caballero, B.H.; Cousins, R.J.; Tucker, K.L.; Ziegler, T.R. Modern Nutrition in Health and Disease, 11th ed.; Wolters Kluwer Health Adis (ESP): Baltimore, MD, USA, 2012; 1616p. [Google Scholar]
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ Clin. Res. Ed. 2012, 346, e7492. [Google Scholar] [CrossRef] [PubMed]
- Fattore, E.; Botta, F.; Agostoni, C.; Bosetti, C. Effects of free sugars on blood pressure and lipids: A systematic review and meta-analysis of nutritional isoenergetic intervention trials. Am. J. Clin. Nutr. 2017, 105, 42–56. [Google Scholar] [CrossRef]
- Prinz, P. The role of dietary sugars in health: Molecular composition or just calories? Eur. J. Clin. Nutr. 2019, 73, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Cuda, S.E.; Censani, M. Pediatric Obesity Algorithm: A Practical Approach to Obesity Diagnosis and Management. Front. Pediatr. 2018, 6, 431. [Google Scholar] [CrossRef]
- World Health Organization. Guideline: Sugars Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Aggarwal, T.; Bhatia, R.C.; Singh, D.; Sobti, P.C. Prevalence of obesity and overweight in affluent adolescents from Ludhiana, Punjab. Indian Pediatr. 2008, 45, 500–502. [Google Scholar] [PubMed]
- Pérez-Elvira, R.; Oltra-Cucarella, J.; Carrobles, J.A.; Moltó, J.; Flórez, M.; Parra, S.; Agudo, M.; Saez, C.; Guarino, S.; Costea, R.M.; et al. Enhancing the Effects of Neurofeedback Training: The Motivational Value of the Reinforcers. Brain Sci. 2021, 11, 457. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Chaix, A.; Panda, S. Daily eating patterns and their impact on health and disease. Trends Endocrinol. Metab. 2016, 27, 69–83. [Google Scholar] [CrossRef]
- Eng, S.; Wagstaff, D.A.; Kranz, S. Eating late in the evening is associated with childhood obesity in some age groups but not in all children: The relationship between time of consumption and body weight status in U.S. children. Int. J. Behav. Nutr. Phys. Act. 2009, 6, 1479–5868. [Google Scholar] [CrossRef]
- Miller, A.L.; Lumeng, J.C.; LeBourgeois, M.K. Sleep patterns and obesity in childhood. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sreevatsava, M.; Narayan, K.M.; Cunningham, S.A. Evidence for interventions to prevent and control obesity among children and adolescents: Its applicability to India. Indian J. Pediatr. 2013, 80 (Suppl. S1), S115–S122. [Google Scholar] [CrossRef]
- Dehghan, M.; Danesh, N.A.; Merchant, A.T. Childhood obesity, prevalence and prevention. Nutr. J. 2005, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Raj, M.; Krishna Kumar, R. Obesity in children and adolescents. Indian J. Med. Res 2010, 132, 598–607. [Google Scholar]
- Gangwisch, J.E.; Malaspina, D.; Boden-Albala, B.; Heymsfield, S.B. Inadequate sleep as a risk factor for obesity: Analyses of the NHANES I. Sleep 2005, 28, 1289–1296. [Google Scholar] [CrossRef]
- Spiegel, K.; Tasali, E.; Penev, P.; Van Cauter, E. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann. Intern. Med. 2004, 141, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Monasta, L.; Batty, G.D.; Cattaneo, A.; Lutje, V.; Ronfani, L.; Van Lenthe, F.J.; Brug, J. Early-life determinants of overweight and obesity: A review of systematic reviews. Obes. Rev. 2010, 11, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Collings, P.J.; Ball, H.L.; Santorelli, G.; West, J.; Barber, S.E.; McEachan, R.R.; Wright, J. Sleep duration and adiposity in early childhood: Evidence for bidirectional associations from the born in Bradford Study. Sleep 2017, 40, zsw054. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.; Hill, C.M.; Harvey, N.C.; Crozier, S.; Robinson, S.M.; Godfrey, K.M.; Cooper, C.; Inskip, H.; SWS Study Group. Duration of sleep at 3 years of age is associated with fat and fat-free mass at 4 years of age: The Southampton Women’s Survey. J. Sleep Res. 2016, 25, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Cespedes, E.M.; Hu, F.B.; Redline, S.; Rosner, B.; Gillman, M.W.; Rifas-Shiman, S.L.; Taveras, E.M. Chronic insufficient sleep and diet quality: Contributors to childhood obesity. Obesity 2016, 24, 184–190. [Google Scholar] [CrossRef]
- Taveras, E.M.; Gillman, M.W.; Pena, M.M.; Redline, S.; Rifas-Shiman, S.L. Chronic sleep curtailment and adiposity. Pediatrics 2014, 133, 1013–1022. [Google Scholar] [CrossRef]
- Bornhorst, C.; Hense, S.; Ahrens, W.; Hebestreit, A.; Reisch, L.; Barba, G.; von Kries, R.; Bayer, O.; IDEFICS Consortium. From sleep duration to childhood obesity—What are the pathways? Eur. J. Pediatr. 2012, 171, 1029–1038. [Google Scholar] [CrossRef]
- Diethelm, K.; Bolzenius, K.; Cheng, G.; Remer, T.; Buyken, A.E. Longitudinal associations between reported sleep duration in early childhood and the development of body mass index, fat mass index and fat free mass index until age 7. Int. J. Pediatr. Obes. 2011, 6, e114–e123. [Google Scholar] [CrossRef] [PubMed]
- Neamțu, B.M.; Visa, G.; Maniu, I.; Ognean, M.L.; Pérez-Elvira, R.; Dragomir, A.; Agudo, M.; Șofariu, C.R.; Gheonea, M.; Pitic, A.; et al. A Decision-Tree Approach to Assist in Forecasting the Outcomes of the Neonatal Brain Injury. Int. J. Environ. Res. Public Health 2021, 18, 4807. [Google Scholar] [CrossRef]
- Paruthi, S.; Brooks, L.J.; D’Ambrosio, C.; Hall, W.A.; Kotagal, S.; Lloyd, R.M.; Malow, B.A.; Maski, K.; Nichols, C.; Quan, S.F.; et al. Recommended amount of sleep for pediatric populations: A consensus statement of the American Academy of Sleep Medicine. J. Clin. Sleep Med. 2016, 12, 785–786. [Google Scholar] [CrossRef]
- Chen, X.; Beydoun, M.A.; Wang, Y. Is sleep duration associated with childhood obesity? A systematic review and meta-analysis. Obesity 2008, 16, 265–274. [Google Scholar] [CrossRef]
- Campbell, M.K. Biological, environmental, and social influences on childhood obesity. Pediatr. Res. 2016, 79, 205–211. [Google Scholar] [CrossRef]
- Lampard, A.M.; Franckle, R.L.; Davison, K.K. Maternal depression and childhood obesity: A systematic review. Prev. Med. 2014, 59, 60–67. [Google Scholar] [CrossRef]
- Rolls, B.J. The Supersizing of America: Portion size and the obesity epidemic. Nutr. Today 2003, 38, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.M.; Aronne, L.J. Causes of obesity. Abdom. Imaging 2012, 37, 730–732. [Google Scholar] [CrossRef]
- James, W.P. The challenge of childhood obesity. Int. J. Pediatr. Obes 2006, 1, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Pombo-Rodrigues, S.; Hashem, K.M.; Tan, M.; Davies, Z.; He, F.J.; MacGregor, G.A. Nutrition Profile of Products with Cartoon Animations on the Packaging: A UK Cross-Sectional Survey of Foods and Drinks. Nutrients 2020, 12, 707. [Google Scholar] [CrossRef]
- Cohen, D.; Babey, S. Candy at the Cash Register—A Risk Factor for Obesity and Chronic Disease. N. Engl. J. Med. 2012, 15, 1381–1383. [Google Scholar] [CrossRef]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac remodeling in obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef]
- Poirier, P.; Bray, G.A.; Giles, T.D.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H.; American Heart Association, & Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: An update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006, 113, 898–918. [Google Scholar] [CrossRef]
- Mathew, B.; Francis, L.; Kaylar, A.; Cone, J. Obesity: Effects on Cardiovascular Disease and its Diagnosis. J. Am. Board Fam. Med. 2008, 21, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M. Obesity in Children [Internet]. 20 February 2019. Available online: https://emedicine.medscape.com/article/985333-overview (accessed on 29 September 2019).
- Llewellyn, A.; Simmonds, M.; Owen, C.G.; Woolacott, N. Childhood obesity as a predictor of morbidity in adulthood: A systematic review and meta-analysis. Obes. Rev. 2016, 17, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Juonala, M.; Magnussen, C.G.; Berenson, G.S.; Venn, A.; Burns, T.L.; Sabin, M.A.; Srinivasan, S.R.; Daniels, S.R.; Davis, P.H.; Chen, W.; et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N. Engl. J. Med. 2011, 365, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.G.; Whincup, P.H.; Orfei, L.; Chou, Q.A.; Rudnicka, A.R.; Wathern, A.K.; Kaye, S.J.; Eriksson, J.G.; Osmond, C.; Cook, D.G. Is body mass index before middle age related to coronary heart disease risk in later life? Evidence from observational studies. Int. J. Obes. 2009, 33, 866–877. [Google Scholar] [CrossRef]
- Kindblom, J.M.; Bygdell, M.; Sondén, A.; Célind, J.; Rosengren, A.; Ohlsson, C. BMI change during puberty and the risk of heart failure. J. Intern. Med. 2018, 283, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, J.S.; Hernesniemi, J.A.; Ruohonen, S.; Hutri-Kähönen, N.; Kähönen, M.; Jokinen, E.; Tossavainen, P.; Kallio, M.; Laitinen, T.; Lehtimäki, T. Influence of early-life body mass index and systolic blood pressure on left ventricle in adulthood—the Cardiovascular Risk in Young Finns Study. Ann. Med. 2021, 53, 160–168. [Google Scholar] [CrossRef]
- Adelborg, K.; Ängquist, L.; Ording, A.; Gjærde, L.K.; Bjerregaard, L.G.; Sørensen, H.T.; Sørensen, T.; Baker, J.L. Levels of and Changes in Childhood Body Mass Index in Relation to Risk of Atrial Fibrillation and Atrial Flutter in Adulthood. Am. J. Epidemiol. 2019, 188, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F.H.; Reisin, E.; Ventura, H.O.; Reisin, E.; Dreslinski, G.R.; Dunn, F.G.; MacPhee, A.A.; Frohlich, E.D. Borderline hypertension and obesity: Two prehypertensive states with elevated cardiac output. Circulation 1982, 66, 55–60. [Google Scholar] [CrossRef]
- Alpert, M.A. Obesity cardiomyopathy: Pathophysiology and evolution of the clinical syndrome. Am. J. Med. Sci. 2001, 321, 225–236. [Google Scholar] [CrossRef]
- Chakko, S.; Allison, M.D.; Mayor, M.; Kessler, K.M.; Materson, B.J.; Myerburg, R.J. Abnormal left ventricular diastolic filling in eccentric left ventricular hypertrophy of obesity. Am. J. Cardiol. 1991, 68, 95–98. [Google Scholar] [CrossRef]
- Lavie, C.J.; Milani, R.V.; Ventura, H.O.; Cardenas, G.A.; Mehra, M.R.; Messerli, F.H. Disparate effects of left ventricular geometry and obesity on mortality in patients with preserved left ventricular ejection fraction. Am. J. Cardiol. 2007, 100, 1460–1464. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C. Childhood obesity and cardiac remodeling: From cardiac structure to myocardial mechanics. J. Cardiovasc. Med. 2015, 16, 538–546. [Google Scholar] [CrossRef]
- Esanu, V.; Palii, I.; Mocanu, V.; Vudu, L.; Esanu, V. Left ventricular remodeling patterns in children with metabolic syndrome. One Health Risk Manag. 2020, 1, 41–49. [Google Scholar] [CrossRef]
- Lavie, C.J.; Amodeo, C.; Ventura, H.O.; Messerli, F.H. Left atrial abnormalities indicating diastolic ventricular dysfunction in cardiopathy of obesity. Chest 1987, 92, 1042–1046. [Google Scholar] [CrossRef]
- De Scheerder, I.; Cuvelier, C.; Verhaaren, R.; De Buyzere, M.; De Backer, G.; Clement, D. Restrictive cardiomyopathy caused by adipositas cordis. Eur. Heart J. 1987, 8, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.M.; García-Espinosa, V.; Curcio, S.; Arana, M.; Chiesa, P.; Giachetto, G.; Zócalo, Y.; Bia, D. Childhood obesity associates haemodynamic and vascular changes that result in increased central aortic pressure with augmented incident and reflected wave components, without changes in peripheral amplification. Int. J. Vasc. Med. 2016, 2016, 3129304. [Google Scholar] [CrossRef] [PubMed]
- Mangner, N.; Scheuermann, K.; Winzer, E.; Wagner, I.; Hoellriegel, R.; Sandri, M.; Zimmer, M.; Mende, M.; Linke, A.; Kiess, W.; et al. Childhood obesity: Impact on cardiac geometry and function. JACC Cardiovasc. Imaging 2014, 7, 1198–1205. [Google Scholar] [CrossRef]
- Genovesi, S.; Antolini, L.; Giussani, M.; Pieruzzi, F.; Galbiati, S.; Valsecchi, M.G.; Brambilla, P.; Stella, A. Usefulness of waist circumference for the identification of childhood hypertension. J. Hypertens. 2008, 26, 1563–1570. [Google Scholar] [CrossRef]
- Faulkner, B. Recent clinical and translational advances in pediatric hypertension. Hypertension 2015, 65, 926–931. [Google Scholar] [CrossRef]
- Zeng, M.; Liang, Y.; Li, H.; Wang, M.; Wang, B.; Chen, X.; Zhou, N.; Cao, D.; Wu, J. Plasma metabolic fingerprinting of childhood obesity by GC/MS in conjunction with multivariate statistical analysis. J. Pharm. Biomed. Anal. 2010, 52, 265–272. [Google Scholar] [CrossRef]
- Urbina, E.M.; Khoury, P.R.; Bazzano, L.; Burns, T.L.; Daniels, S.; Dwyer, T.; Hu, T.; Jacobs, D.R., Jr.; Juonala, M.; Prineas, R.; et al. Relation of Blood Pressure in Childhood to Self-Reported Hypertension in Adulthood. Hypertension 2019, 73, 1224–1230. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y. Tracking of blood pressure from childhood to adulthood: A systematic review and meta-regression analysis. Circulation 2008, 117, 3171–3180. [Google Scholar] [CrossRef] [PubMed]
- Marcon, D.; Tagetti, A.; Fava, C. Subclinical Organ Damage in Children and Adolescents with Hypertension: Current Guidelines and Beyond. High Blood Press Cardiovasc. Prev. 2019, 26, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, R.; de Sousa, G.; Toschke, A.M.; Reinehr, T. Intima-media thickness in obese children before and after weight loss. Pediatrics 2006, 118, 2334–2340. [Google Scholar] [CrossRef]
- Weberruß, H.; Böhm, B.; Pirzer, R.; Böhm, B.; Pozza, R.D.; Netz, H.; Oberhoffer, R. Intima-media thickness and arterial function in obese and non-obese children. BMC Obes. 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Nunez, F.; Martinez-Costa, C.; Sanchez-Zahonero, J.; Morata, J.; Chorro, F.J.; Brines, J. Carotid artery stiffness as an early marker of vascular lesions in children and adolescents with cardiovascular risk factors. Rev. Esp. Cardiol. 2010, 63, 1253–1260. [Google Scholar] [CrossRef]
- Lurbe, E.; Agabiti-Rosei, E.; Cruickshank, J.K.; Dominiczak, A.; Erdine, S.; Hirth, A.; Invitti, C.; Litwin, M.; Mancia, G.; Pall, D.; et al. 2016 European Society of Hypertension guidelines for the management of high blood pressure in children and adolescents. J. Hypertens. 2016, 34, 1887–1920. [Google Scholar] [CrossRef]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P., 3rd; Tracy, R.E.; Wattigney, W.A. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults: The Bogalusa Heart Study. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Milei, J.; Lavezzi, A.M.; Ottaviani, G.; Grana, D.R.; Stella, I.; Matturri, L. Perinatal and infant early atherosclerotic coronary lesions. Can. J. Cardiol. 2008, 24, 137–141. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Ayer, J.G.J. Childhood risk factors for adult cardiovascular disease and primary prevention in childhood. Heart 2006, 92, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lerman, L.O.; de Nigris, F.; Gossl, M.; Balestrieri, M.L.; Lerman, A. Rethinking primary prevention of atherosclerosis-related diseases. Circulation 2006, 114, 2517–2527. [Google Scholar] [CrossRef]
- Lovren, F.; Teoh, H.; Verma, S. Obesity and atherosclerosis: Mechanistic insights. Can. J. Cardiol. 2015, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Olsen, L.W.; Sorensen, T.I. Childhood body-mass index and the risk of coronary heart disease in adulthood. N. Engl. J. Med. 2007, 357, 2329–2337. [Google Scholar] [CrossRef]
- Daniels, S.R. Diet and primordial prevention of cardiovascular disease in children and adolescents. Circulation 2007, 116, 973–974. [Google Scholar] [CrossRef][Green Version]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Genovesi, S.; Giussani, M.; Orlando, A.; Battaglino, M.G.; Nava, E.; Parati, G. Prevention of cardiovascular diseases in children and adolescents. High Blood Press Cardiovasc. Prev. 2019, 26, 191–197. [Google Scholar] [CrossRef]
- Tirosh, A.; Afek, A.; Shai, I.; Dubnov-Raz, G.; Ayalon, N.; Gordon, B.; Derazne, E.; Tzur, D.; Shamis, A.; Vinker, S.; et al. Adolescent BMI trajectory and risk of diabetes versus coronary disease. N. Engl. J. Med. 2011, 364, 1315–1325. [Google Scholar] [CrossRef]
- National Heart, Lung, and Blood Institute; US Department of Health and Human Service; National Institutes of Health. Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents, Summary Report. Pediatrics 2011, 128 (Suppl. S5), S213. [Google Scholar] [CrossRef]
- Cook, S.; Kavey, R.E. Dyslipidemia and pediatric obesity. Pediatr. Clin. N. Am. 2011, 58, 1363–1373. [Google Scholar] [CrossRef]
- Sánchez, J.; Priego, T.; Picó, C.; Ahrens, W.; De Henauw, S.; Fraterman, A.; Mårild, S.; Molnár, D.; Moreno, L.A.; Peplies, J.; et al. Blood cells as a source of transcriptional biomarkers of childhood obesity and its related metabolic alterations: Results of the IDEFICS study. J. Clin. Endocrinol. Metab. 2012, 97, 648–652. [Google Scholar] [CrossRef]
- MicroRNAs-Oses, M.; Margareto Sanchez, J.; Portillo, M.P.; Aguilera, C.M.; Labayen, I. Circulating miRNAs as Biomarkers of Obesity and Obesity-Associated Comorbidities in Children and Adolescents: A Systematic Review. Nutrients 2019, 11, 2890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.L.; Yang, Q.M.; Li, C.; Chen, J.; Xiang, M.; Chen, M.M.; Yan, M.; Zhu, Z.G. Identification of biomarkers for childhood obesity based on expressional correlation and functional similarity. Mol. Med. Rep. 2018, 17, 109–116. [Google Scholar] [CrossRef]
- Stolzman, S.; Bement, M.H. Inflammatory Markers in Pediatric Obesity: Health and Physical Activity Implications. Infant Child. Adolesc. Nutr. 2012, 4, 297–302. [Google Scholar] [CrossRef][Green Version]
- Prats-Puig, A.; Gispert-Saüch, M.; Díaz-Roldán, F.; Carreras-Badosa, G.; Osiniri, I.; Planella-Colomer, M.; Mayol, L.; de Zegher, F.; Ibánez, L.; Bassols, J.; et al. Neutrophil-to-lymphocyte ratio: An inflammation marker related to cardiovascular risk in children. Thromb. Haemost. 2015, 114, 727–734. [Google Scholar] [CrossRef]
- Aydin, M.; Yilmaz, A.; Donma, M.M.; Tulubas, F.; Demirkol, M.; Erdogan, M.; Gurel, A. Neutrophil/lymphocyte ratio in obese adolescents. North Clin. Istanb. 2015, 2, 87–91. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buyukkaya, E.; Karakas, M.F.; Karakas, E.; Akcay, A.B.; Tanboga, I.H.; Kurt, M.; Sen, N. Correlation of neutrophil to lymphocyte ratio with the presence and severity of metabolic syndrome. Clin. Appl. Thromb. Hemost. 2014, 20, 159–163. [Google Scholar] [CrossRef]
- Bhat, T.; Teli, S.; Rijal, J.; Bhat, H.; Raza, M.; Khoueiry, G.; Meghani, M.; Akhtar, M.; Costantino, T. Neutrophil to lymphocyte ratio and cardiovascular diseases: A review. Expert Rev. Cardiovasc. Ther. 2013, 11, 55–59. [Google Scholar] [CrossRef]
- Furuncuoğlu, Y.; Tulgar, S.; Dogan, A.N.; Cakar, S.; Tulgar, Y.K.; Cakiroglu, B. How obesity affects the neutrophil/lymphocyte and platelet/lymphocyte ratio, systemic immune-inflammatory index and platelet indices: A retrospective study. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1300–1306. [Google Scholar] [PubMed]
- Sacheck, J. Pediatric obesity: An inflammatory condition? J. Parenter. Enter Nutr. 2008, 32, 633–637. [Google Scholar] [CrossRef]
- Venner, A.A.; Lyon, M.E.; Doyle-Baker, P.K. Leptin: A potential biomarker for childhood obesity? Clin. Biochem. 2006, 39, 1047–1056. [Google Scholar] [CrossRef]
- Kralisch, S.; Fasshauer, M. Adipocyte fatty acid binding protein: A novel adipokine involved in the pathogenesis of metabolic and vascular disease? Diabetologia 2013, 56, 10–21. [Google Scholar] [CrossRef]
- Cambuli, V.M.; Musiu, M.C.; Incani, M.; Paderi, M.; Serpe, R.; Marras, V.; Cossu, E.; Cavallo, M.G.; Mariotti, S.; Loche, S.; et al. Assessment of adiponectin and leptin as biomarkers of positive metabolic outcomes after lifestyle intervention in overweight and obese children. J. Clin. Endocrinol. Metab. 2008, 93, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Niroumand, S.; Khajedaluee, M.; Khadem-Rezaiyan, M.; Abrishami, M.; Juya, M.; Khodaee, G.; Dadgarmoghaddam, M. Atherogenic Index of Plasma (AIP): A marker of cardiovascular disease. Med. J. Islam. Repub. Iran. 2015, 29, 240. [Google Scholar]
- Zhu, X.; Yu, L.; Zhou, H.; Ma, Q.; Zhou, X.; Lei, T.; Hu, J.; Xu, W.; Yi, N.; Lei, S. Atherogenic index of plasma is a novel and better biomarker associated with obesity: A population-based cross-sectional study in China. Lipids Health Dis. 2018, 17, 37. [Google Scholar] [CrossRef]
- Dobiášová, M. Atherogenic index of plasma [log(triglycerides/HDL-cholesterol)]: Theoretical and practical implications. Clin. Chem. 2004, 50, 1113–1115. [Google Scholar] [CrossRef]
- Burgert, T.S.; Taksali, S.E.; Dziura, J.; Goodman, T.R.; Yeckel, C.W.; Papademetris, X.; Constable, R.T.; Weiss, R.; Tamborlane, W.V.; Savoye, M.; et al. Alanine aminotransferase levels and fatty liver in childhood obesity: Associations with insulin resistance, adiponectin, and visceral fat. J. Clin. Endocrinol. Metab. 2006, 91, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Labayen, I.; Ruiz, J.R.; Ortega, F.B.; Davis, C.L.; Rodríguez, G.; González-Gross, M.; Breidenassel, C.; Dallongeville, J.; Marcos, A.; Widhalm, K.; et al. Liver enzymes and clustering cardiometabolic risk factors in European adolescents: The HELENA study. Pediatr. Obes. 2015, 10, 361–370. [Google Scholar] [CrossRef] [PubMed]













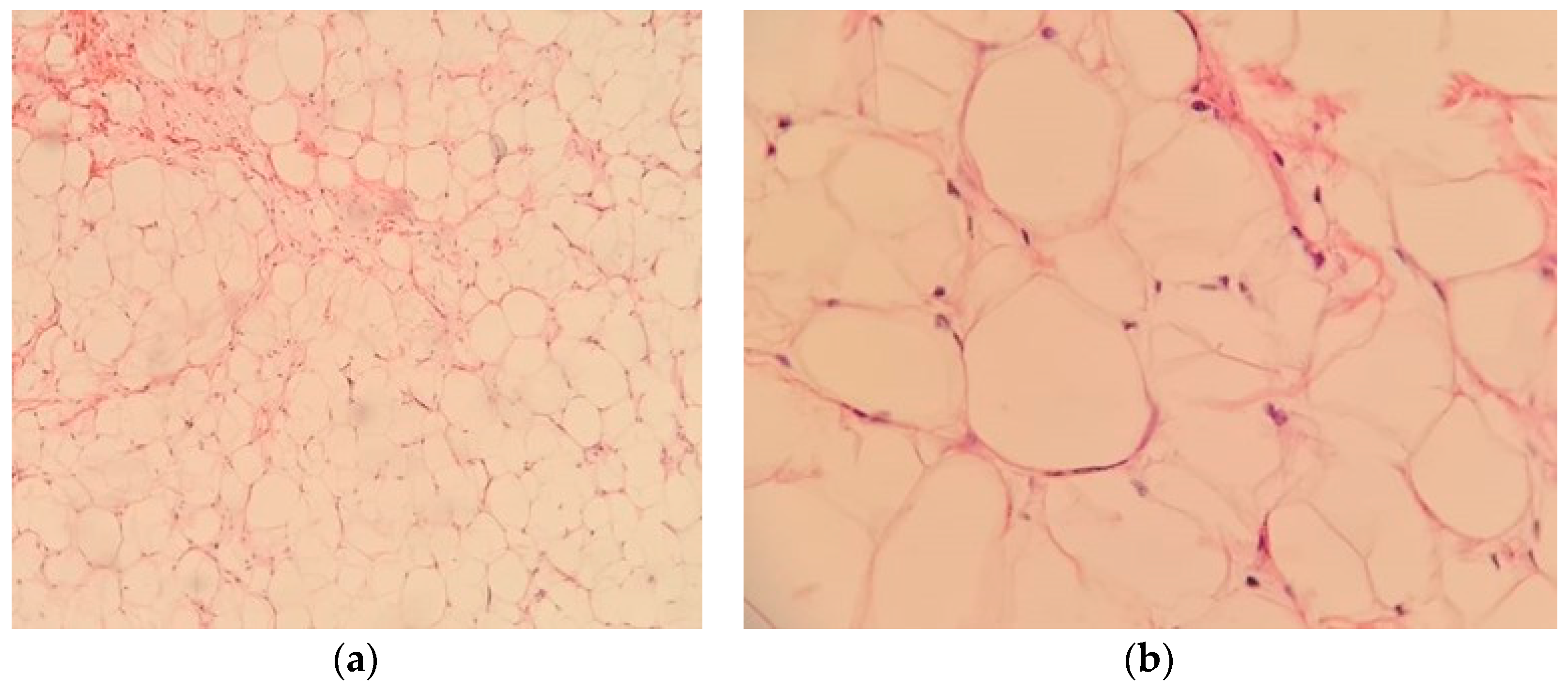{kind=link}
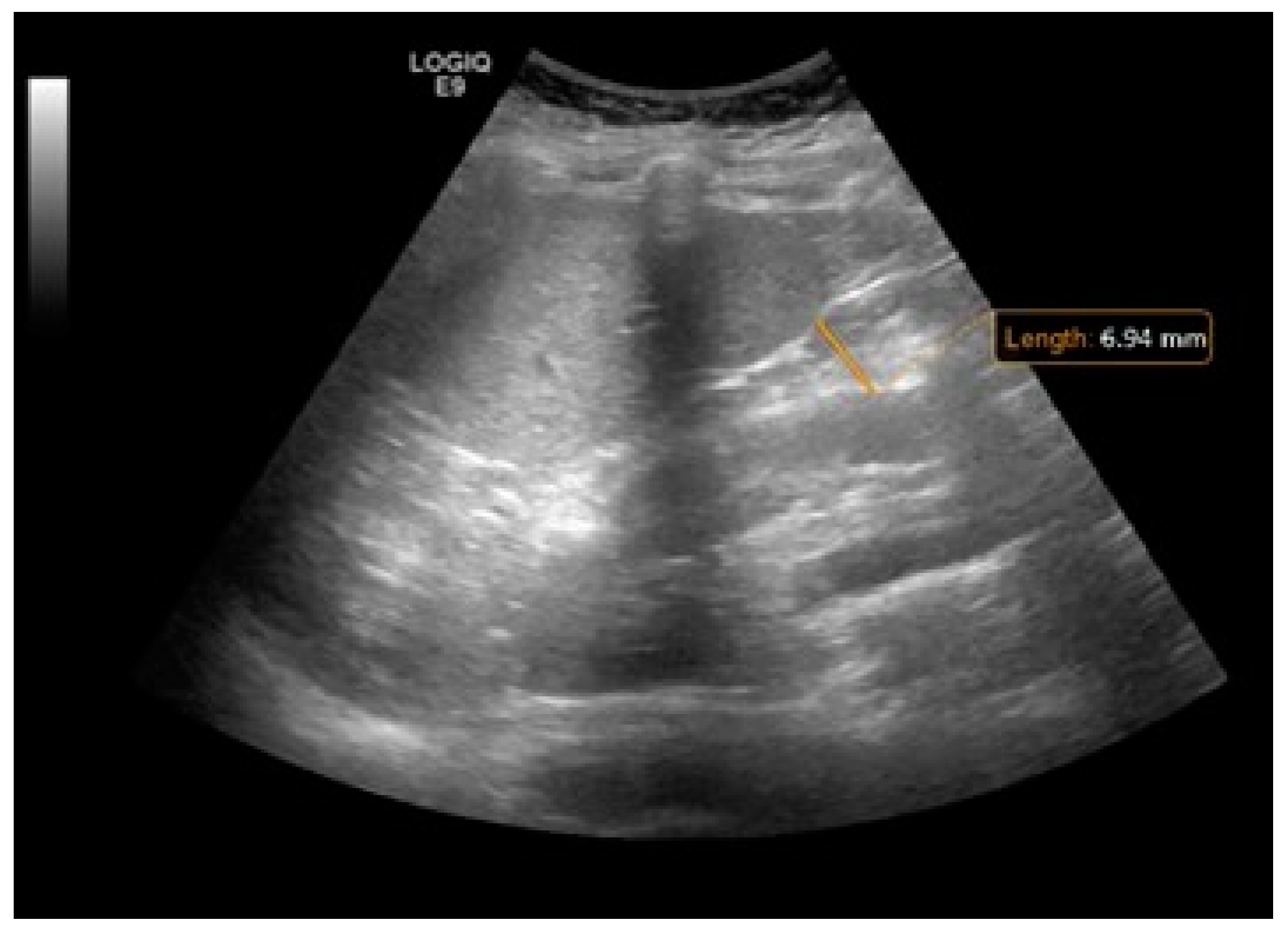{kind=link}
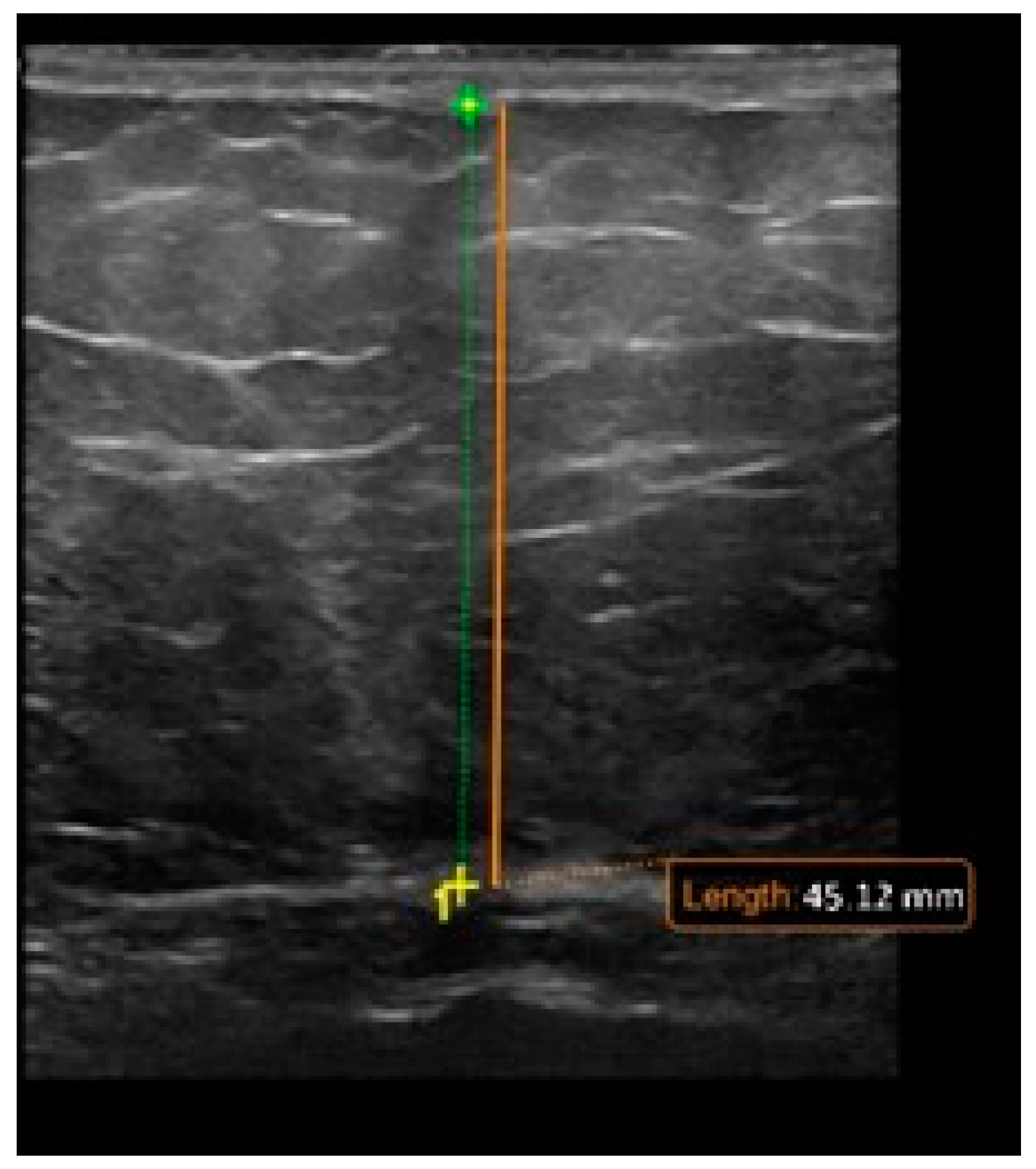{kind=link}
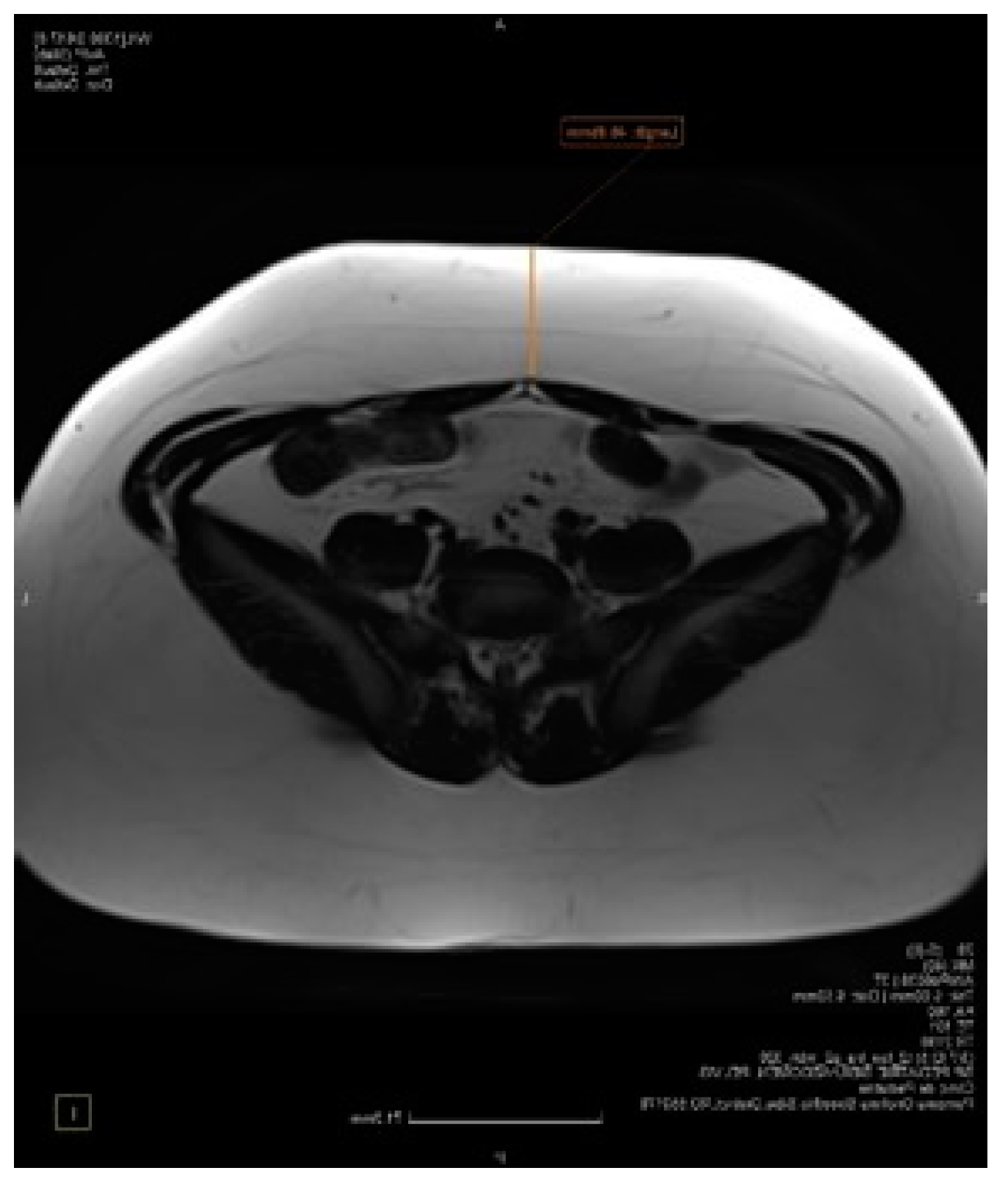{kind=link}
{kind=link}
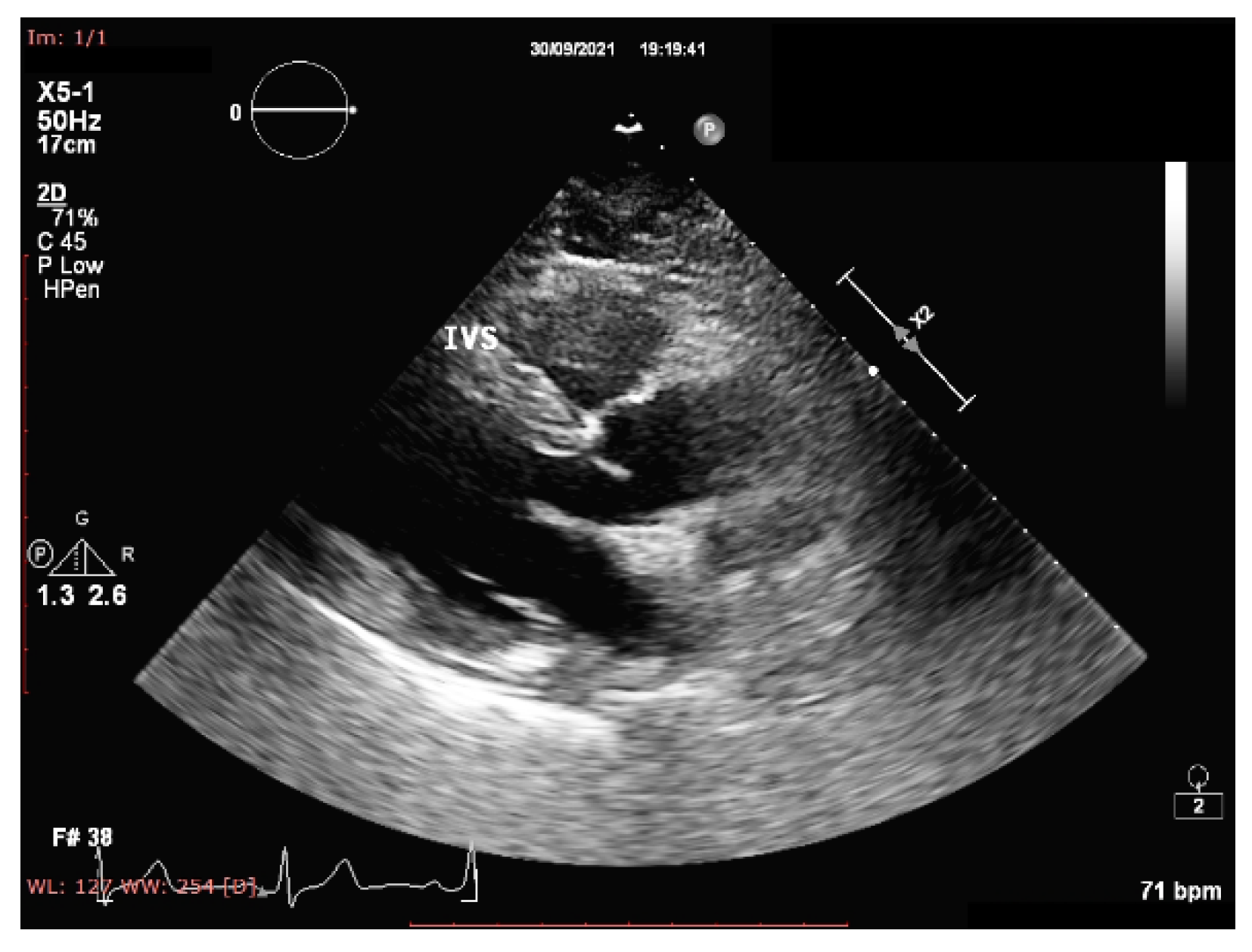{kind=link}
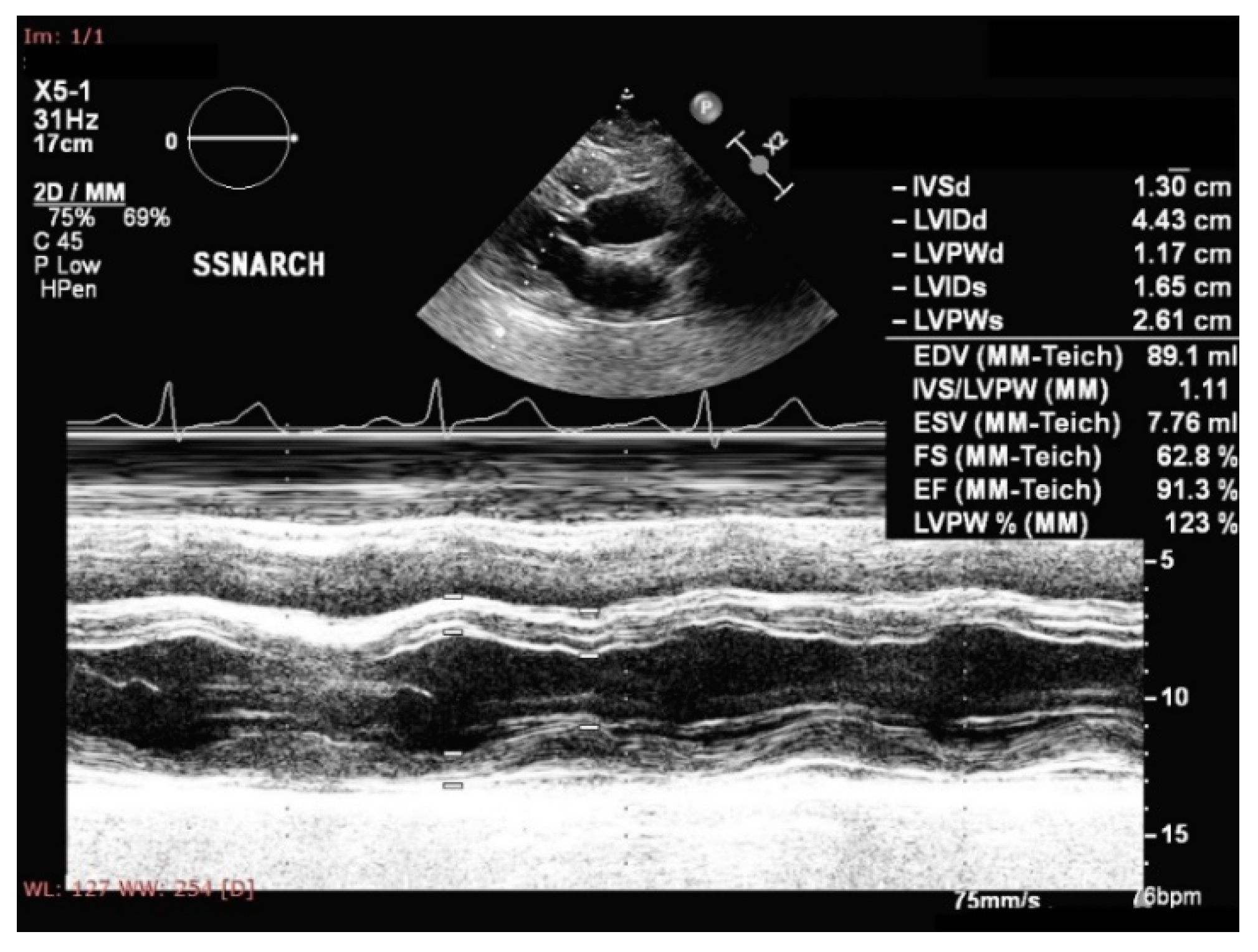{kind=link}
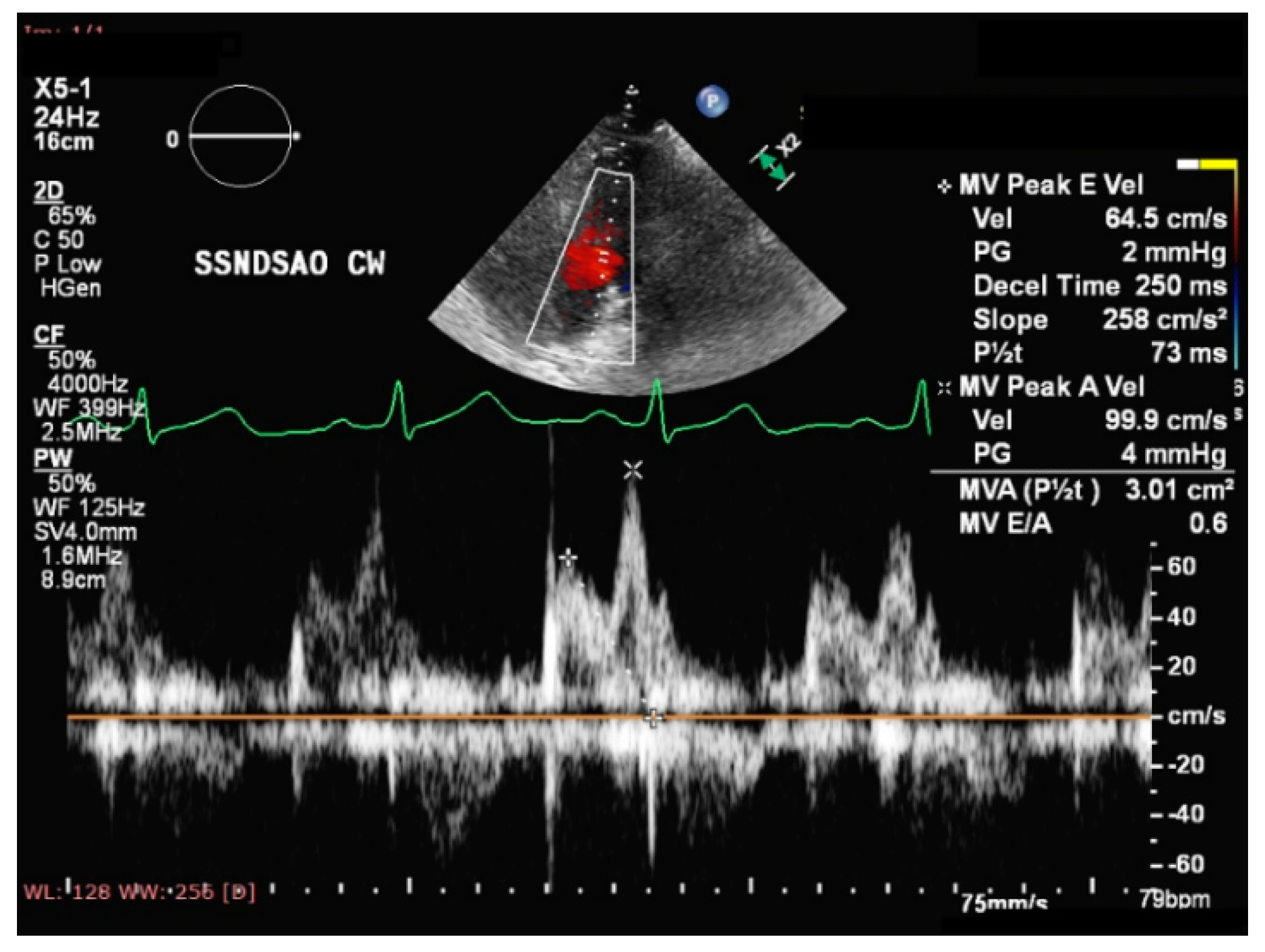{kind=link}
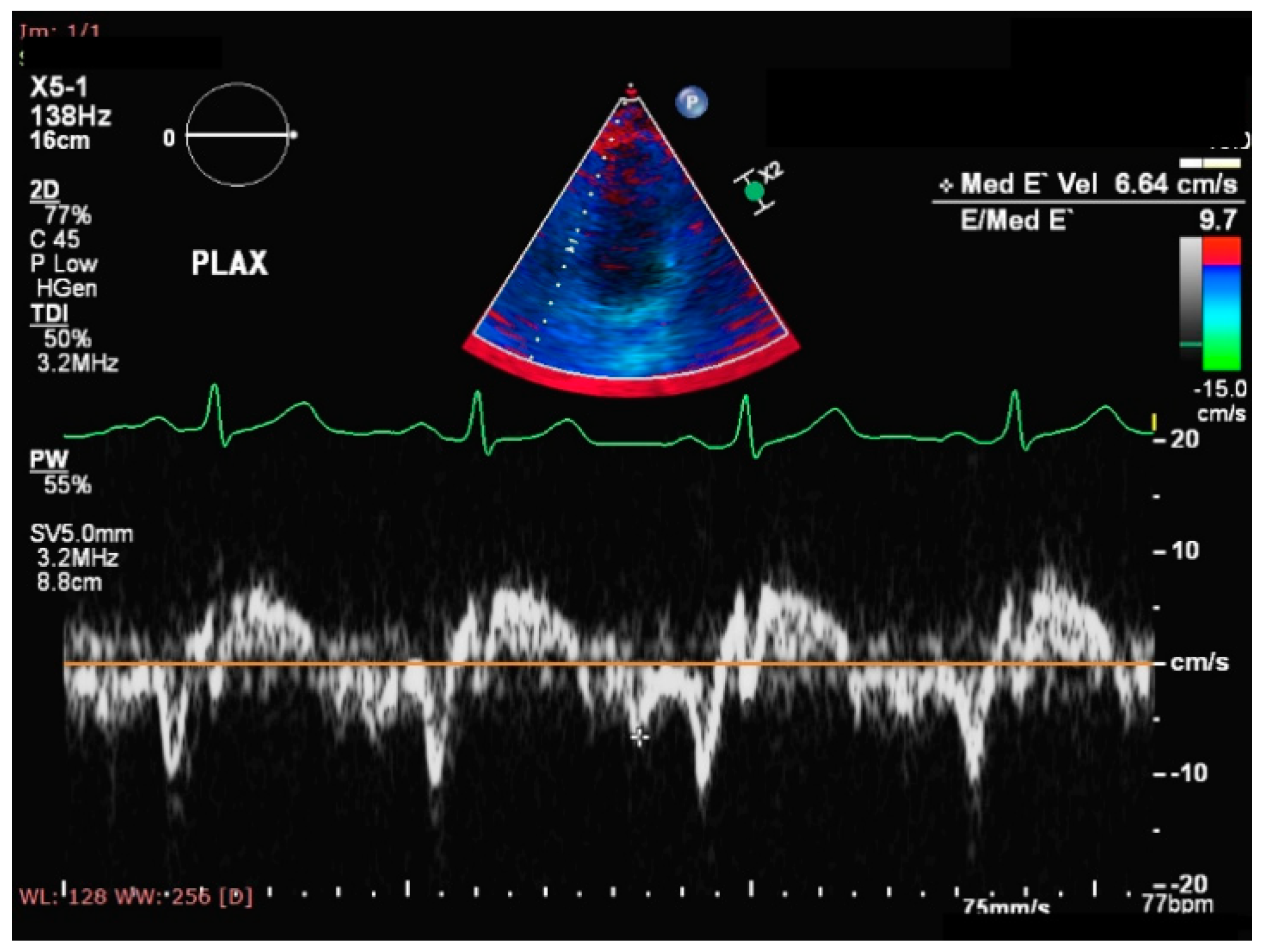{kind=link}
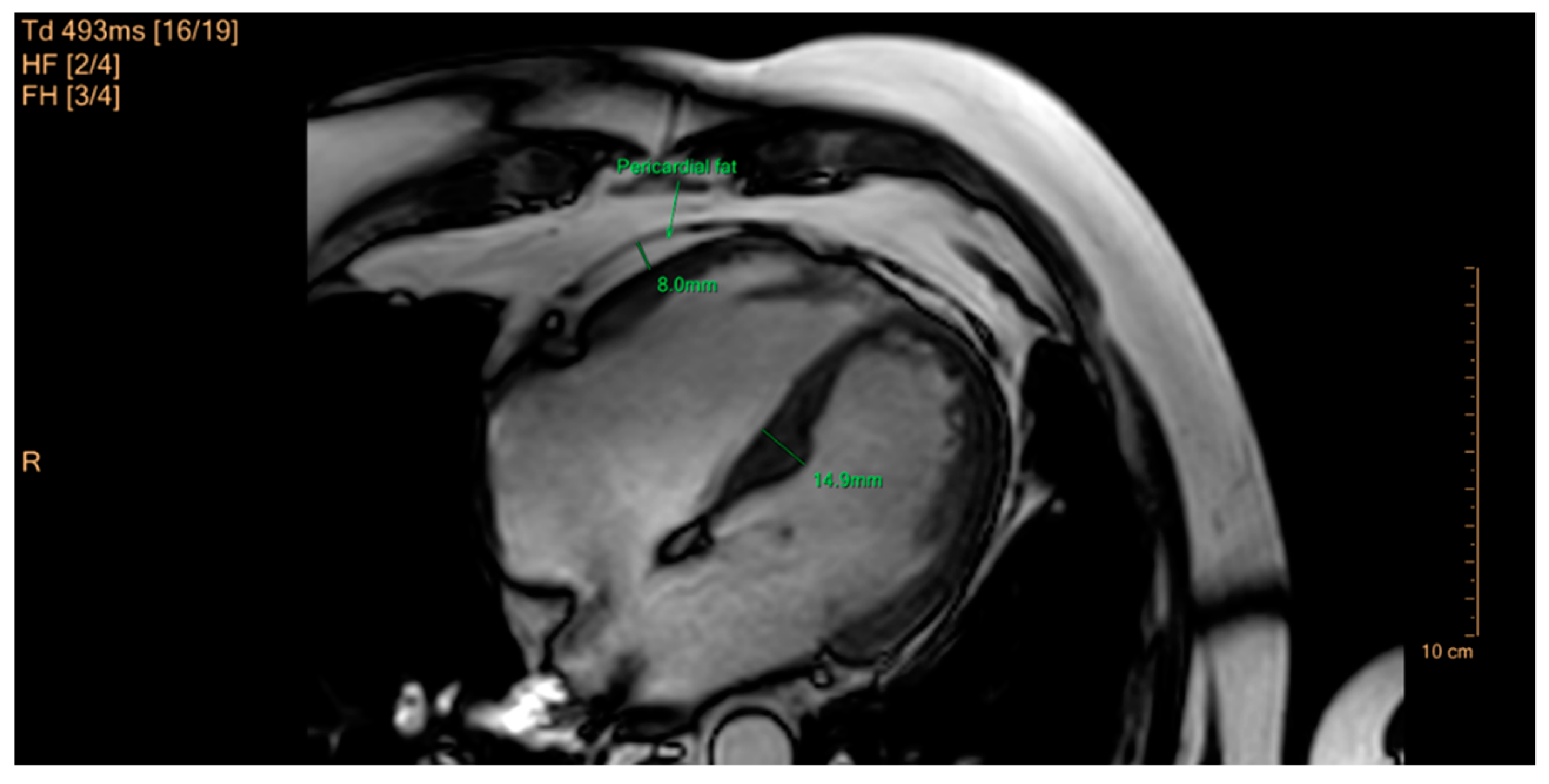{kind=link}
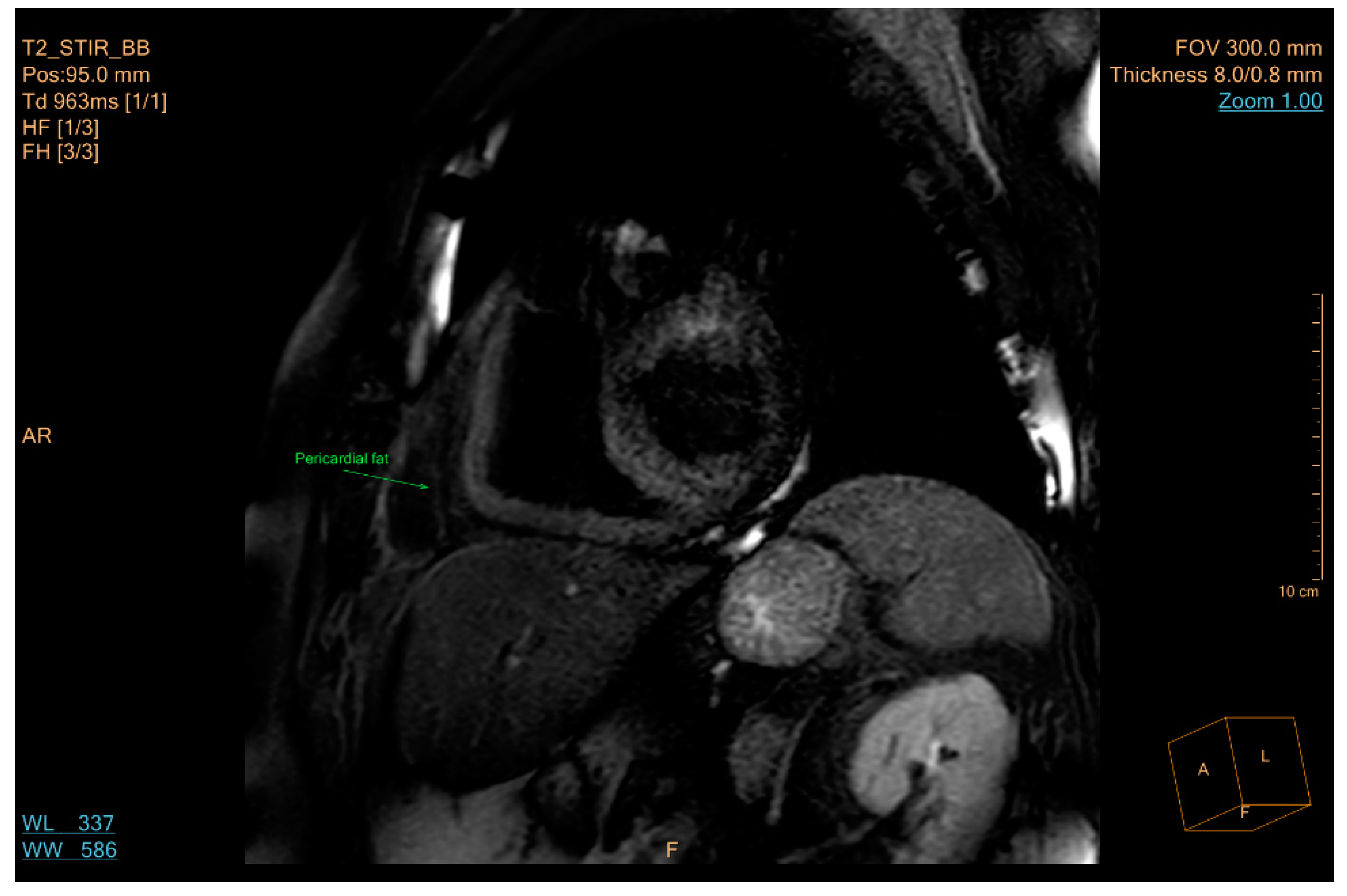{kind=link}
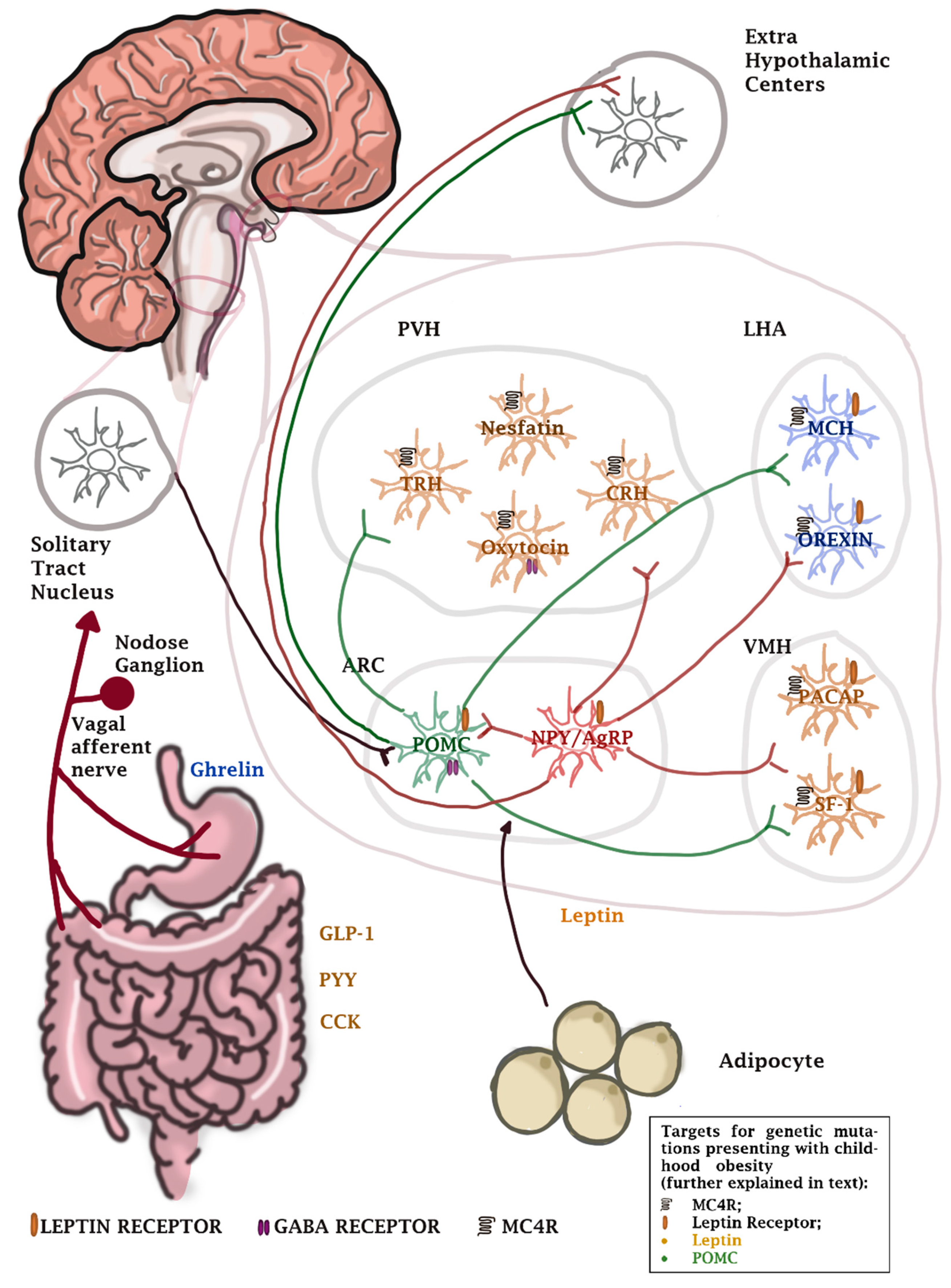{kind=link}
| Location | Year | Type of Weight Excess | Age Group (Years) | Prevalence | References |
|---|---|---|---|---|---|
| United States of America | 2012 | Overweight or obesity | 2–5 | 22.8% | [19] |
| 6–11 | 34.2% | ||||
| 12–19 | 34.5% | ||||
| Obesity | 2–5 | 8.4% | |||
| 6–11 | 17.7% | ||||
| 12–19 | 20.5% | ||||
| Latin America | 2008–2013 | Overweight | 0–5 | 7.1% | [20] |
| Overweight or obesity | 6–11 | 18.9–36.9% | |||
| 12–19 | 16.6–35.8% | ||||
| Asia | 1999–2017 | Overweight | 6–11 | 11.2% | [21] |
| 12–19 | 14,6% | ||||
| Obesity | 6–11 | 5.8% | |||
| 12–19 | 8.6% | ||||
| Africa | 2017 | Overweight and obesity | 0–5 | 8–16% | [22] |
| Australia | 2014–2015 | Overweight | 2–4 | 11% | [23] |
| 5–17 | 20% | ||||
| Obesity | 2–4 | 9% | |||
| 5–17 | 7% | ||||
| Europe | 2011–2016 | Overweight and obesity | 2–13 | 21.3% | [24] |
| Obesity | 5.7% |
| Method | Functioning Principle | Advantages | Disadvantages | Ref |
|---|---|---|---|---|
| Dual-energy X-Ray absorptiometry | Variable X-Ray absorption of different tissues | Proven accuracy in animal studies | Use of algorithms not tailored to pediatric populations Unsatisfactory reproducibility X-Ray exposure | [100,101,102,103] |
| Bioelectrical impedance analysis | Variable electrical impedance of different tissues, in accordance with different water content | Non-invasive | Error susceptibility due to the approximation of the water content of each tissue Use of algorithms not tailored to pediatric populations Cumbersome protocol Imprecise results for extreme values of the determined parameter | [104,105,106,107,108] |
| Hydrostatic weighing | Variable density of different tissues, determined by comparison with the density of water | Non-invasive | Error susceptibility due to the approximation of the density of different tissues which can be particularly variable in pediatric patientsProblematic adherence to measuring protocol of pediatric patients | [109,110,111,112] |
| Air displacement plethysmography | Determining body density by measuring different parameters obtained during a series of thermodynamic processes. | Non-invasive Very good adherence to measurement protocol Can be used even in newborns and infants | High cost Error susceptibility due to the approximation of the density of different tissues Error susceptibility due to approximations regarding the thermodynamic processes involved | [113,114,115,116,117,118,119,120,121] |
| Stable isotope dilution techniques | Calculating total body water based on the ingestion of stable isotopes with uniform distribution within the body and the variable water content of different tissues | Non-invasive Relatively low cost No adverse effects documented yet | Error susceptibility due to the approximation of the water content of different tissues | [122,123] |
| Method | Functioning Principle | Advantages | Disadvantages | Ref |
|---|---|---|---|---|
| Ultrasound | Reflection of ultrasound waves at the interface between tissues of different densities Measurement of subcutaneous adipose tissue thickness and approximation of visceral adipose burden based on the thickness of preperitoneal fat | Non-invasive Readily accessible | Operator-dependence Lack of standardized measurement protocol Insufficient data on pediatric patients | [124,125,126,127,128] |
| Computerized Tomography | Varying absorption of X-rays in different tissuesSectional imaging and 3D reconstruction | High accuracy | Contraindicated in pediatric patients for adiposity evaluation due to high X-Ray exposure | [129,130] |
| Magnetic Resonance Imaging | Sectional imaging technique based on the behavior of protons under the influence of a variable high-intensity electromagnetic field | High accuracy Non-invasive | High cost | [131,132,133,134,135,136,137,138,139,140,141] |
| Reference | Obesity Parameter | Investigated Association | Subject Ages (Childhood) | Subject Ages (Adulthood) | Parameter | Result (95%CI) | |
|---|---|---|---|---|---|---|---|
| Llewellyn et al. * [283] | BMI | Diabetes type 2 | 6 and under | 19–73 | OR/StdBMI | 1.23 (1.10–1.37) | |
| 7 to 11 | 1.78 (1.51–2.10) | ||||||
| 12 to 18 | 1.70 (1.30–2.22) | ||||||
| Coronary heart disease | 7 to 11 | 1.14 (1.08–1.21) | |||||
| 12 to 18 | 1.30 (1.16–1.47) | ||||||
| Hypertension | 7 to 11 | 1.67 (0.89–3.13) | |||||
| 12 to 18 | 1.29 (1.19–1.40) | ||||||
| Juonala et al. *[284] | BMI | Diabetes type 2 | 3 to 19 | 23–46 | RR (O) | 2.4 (1.6–3.6) | |
| Hypertension | 1.8 (1.5–2.1) | ||||||
| High-risk LDL cholesterol | 1.4 (1.2–1.8) | ||||||
| High-risk HDL cholesterol | 1.4 (1.2–1.6) | ||||||
| High risk triglycerides | 1.6 (1.3–1.9) | ||||||
| Owen et al. * [285] | BMI | Coronary heart disease | 7 to 18 | 25–77 | OR/StdBMI | 1.09 (1–1.20) | |
| Kindblom et al. [286] | BMI | Heart failure | 8 vs. 20 | Mean FUP = 37.7yrs after age 20 | HR(Nw/O) | 3.14 (2.25–4.38) | |
| HR(O/O) | 2.85 (1.83–4.45) | ||||||
| Heiskanen et al. [287] | BMI | Eccentric LV hypertrophy | 6 to 18 | 34–49 | OR(O/Ob) | 2.04 (1.35–3.07) | |
| Adelborg et al. [288] | BMI (>90th percentile of study population) | Atrial fibrillation/flutter | 7 | Boys | >25 | HR | 1.35 (1.25–1.45) |
| Girls | 1.26 (1.14–1.38) | ||||||
| 10 | Boys | 1.42 (1.32–1.53) | |||||
| Girls | 1.32 (1.20–1.45) | ||||||
| 13 | Boys | 1.46 (1.36–1.56) | |||||
| Girls | 1.38 (1.27–1.51) | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negrea, M.O.; Neamtu, B.; Dobrotă, I.; Sofariu, C.R.; Crisan, R.M.; Ciprian, B.I.; Domnariu, C.D.; Teodoru, M. Causative Mechanisms of Childhood and Adolescent Obesity Leading to Adult Cardiometabolic Disease: A Literature Review. Appl. Sci. 2021, 11, 11565. https://doi.org/10.3390/app112311565
Negrea MO, Neamtu B, Dobrotă I, Sofariu CR, Crisan RM, Ciprian BI, Domnariu CD, Teodoru M. Causative Mechanisms of Childhood and Adolescent Obesity Leading to Adult Cardiometabolic Disease: A Literature Review. Applied Sciences. 2021; 11(23):11565. https://doi.org/10.3390/app112311565
Chicago/Turabian StyleNegrea, Mihai Octavian, Bogdan Neamtu, Ioana Dobrotă, Ciprian Radu Sofariu, Roxana Mihaela Crisan, Bacila Ionut Ciprian, Carmen Daniela Domnariu, and Minodora Teodoru. 2021. "Causative Mechanisms of Childhood and Adolescent Obesity Leading to Adult Cardiometabolic Disease: A Literature Review" Applied Sciences 11, no. 23: 11565. https://doi.org/10.3390/app112311565
APA StyleNegrea, M. O., Neamtu, B., Dobrotă, I., Sofariu, C. R., Crisan, R. M., Ciprian, B. I., Domnariu, C. D., & Teodoru, M. (2021). Causative Mechanisms of Childhood and Adolescent Obesity Leading to Adult Cardiometabolic Disease: A Literature Review. Applied Sciences, 11(23), 11565. https://doi.org/10.3390/app112311565