
Strategies for Post-Translational Control of Protein Expression and Their Applications



Abstract
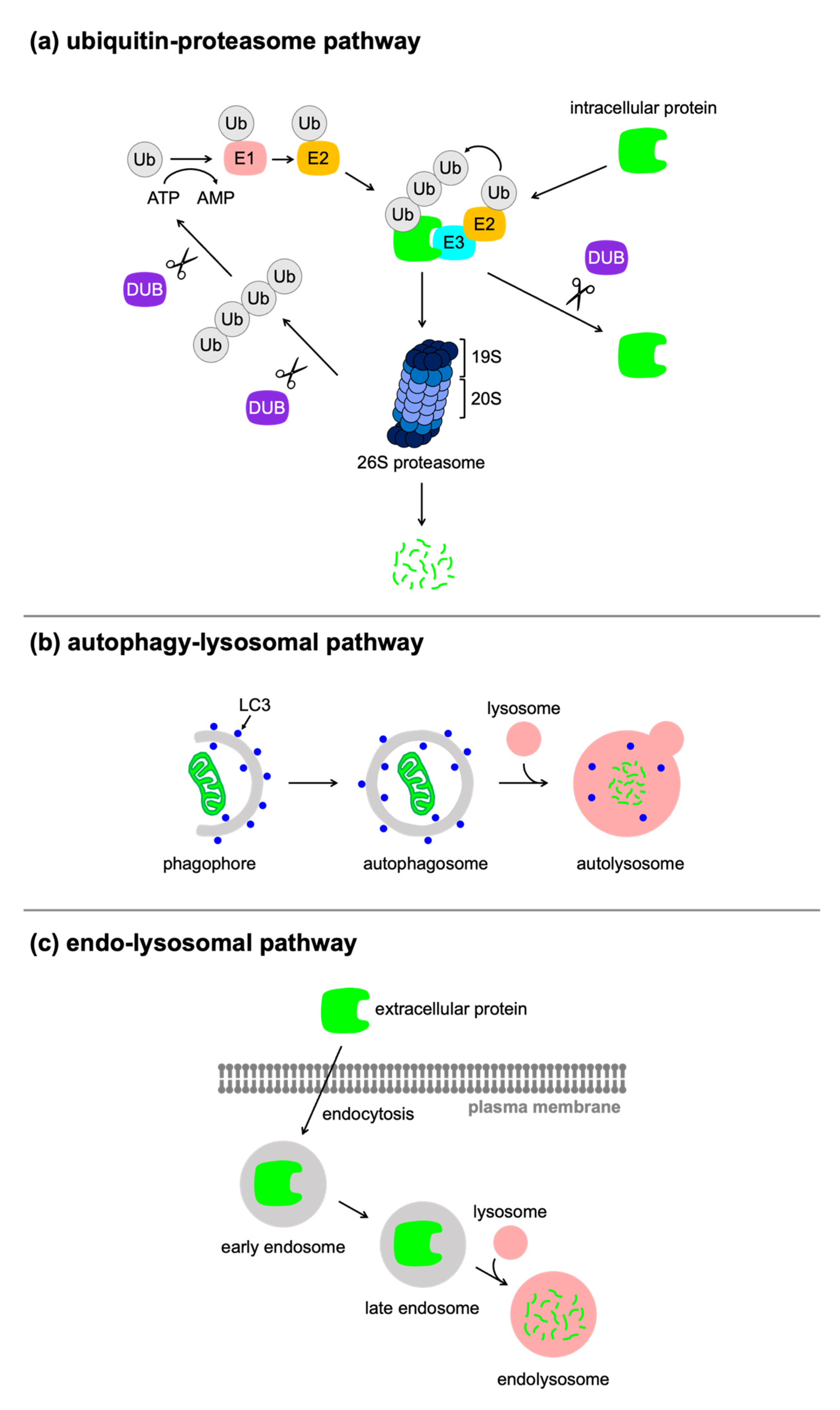
:1. Introduction
2. Methodology
3. Targeted Protein Degradation Using Bifunctional Molecules or Antibodies
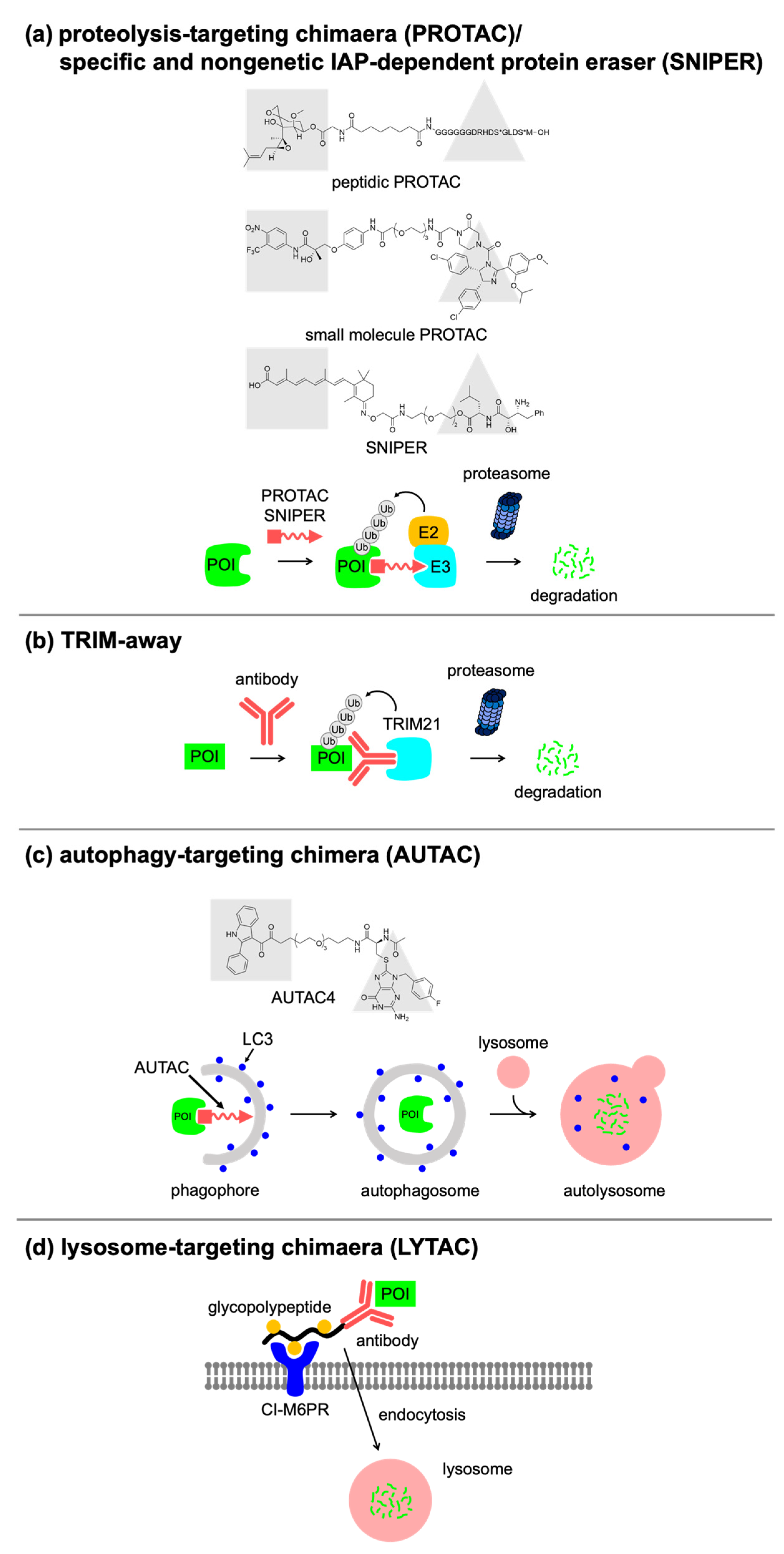
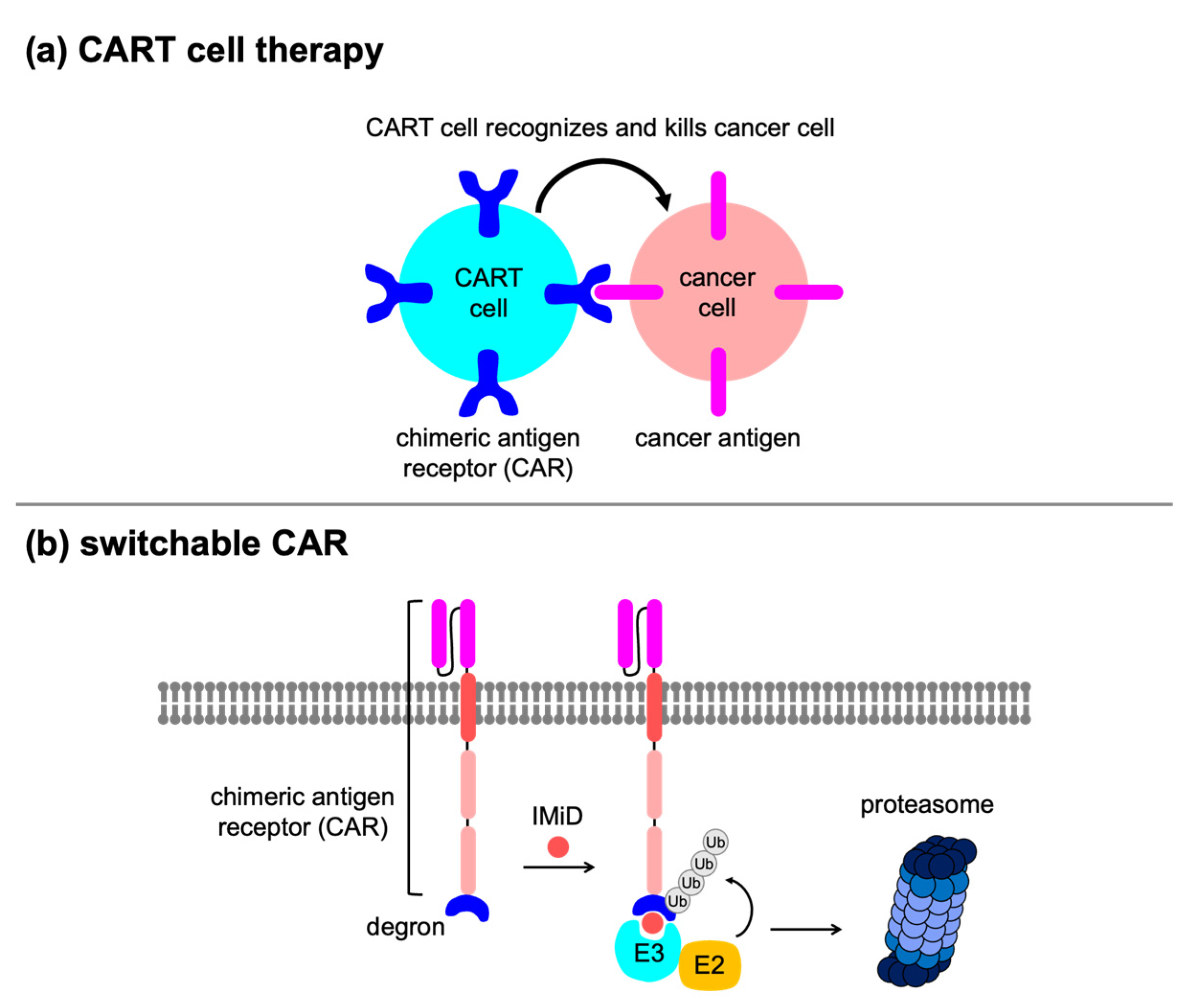
3.1. PROTAC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Proteolytic Pathway | Feature | References |
|---|---|---|---|
| PROTAC/SNIPER | Ubiquitin–proteasome pathway | Originally developed bifunctional molecules | [21,22,23] |
| CLIPTAC | Ubiquitin–proteasome pathway | In-cell generation of bifunctional molecule by the biorthogonal reaction | [25] |
| opto-PROTAC | Ubiquitin–proteasome pathway | Light-inducible switch on PROTAC by photolabile caging group | [26] |
| PHOTAC | Ubiquitin–proteasome pathway | Light-inducible switch on PROTAC by photo-reversible isomerization of azobenzene moiety | [27] |
| AbTAC | Ubiquitin–proteasome pathway | Capable of degrading the cell-surface protein | [28] |
| Antibody-PROTAC conjugate | Ubiquitin–proteasome pathway | Cell-type selective targeting | [29] |
| TRIM away | Ubiquitin–proteasome pathway | Selective degradation of post-translational modified protein and mutant variants | [30,31] |
| AUTAC | Autophagy-lysosome pathway | Capable of targeting cellular organelles | [32] |
| ATTEC | Autophagy-lysosome pathway | Molecular glue type degrader for targeting abnormal proteins | [33,34] |
| LYTAC | Endocytosis | Capable of targeting extracellular and membrane-bound protein | [35,36] |
3.2. Antibody-Based Approaches
3.3. Autophagy/Lysosome-Based Approaches
4. Conditional Control of Protein Stability Using Genetic Manipulation
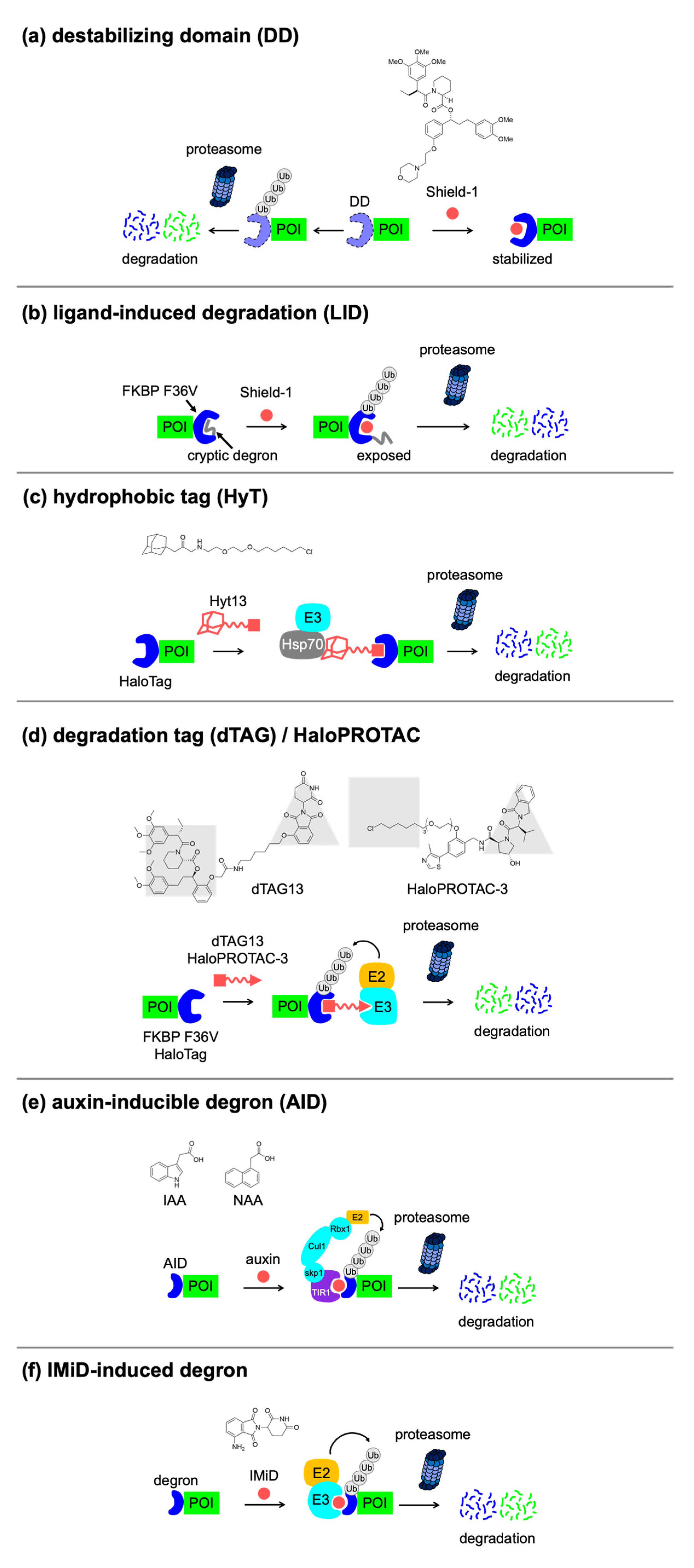
4.1. Small-Molecule-Switchable Degrons
4.2. Light-Switchable Degrons
4.3. Nanobody-Based Degrons
| Degron Technology | Tag Size | Switch | Number of Component(s) | Features | References |
|---|---|---|---|---|---|
| DD | FKBP DD (12 kDa), DHFR DD (18 kDa), etc. | Shield-1 (FKBP DD), TMP (DHFR DD), etc. | 1 | Drug-induced protein stabilization | [38,39,40,41] |
| LID | FKBP F36V-degron (13 kDa) | Shield-1 | 1 | Drug-induced protein degradation | [48] |
| HaloPROTAC | Halo tag (34 kDa) | HaloPROTAC-3 | 1 | Drug-induced protein degradation | [49] |
| HyT | Halo tag (34 kDa) | Hyt13 | 1 | Drug-induced protein degradation | [50] |
| dTAG | FKBP F36V (12 kDa) | dTAG13 | 1 | Drug-induced protein degradation | [53] |
| AID | AID/IAA (25 kDa), mAID (7 kDa) | Auxin (IAA, NAA) | 2 | Drug-induced protein degradation | [54] |
| IMiD-induced degron | IKZF3-based degron (6 kDa), SALL4 degron (3 kDa) | IMiD (thalidomide, pomalidomide, 5-hydroxythalidomide) | 1 | Drug-induced protein degradation | [62,63] |
| Photosensitive degron/B-LID | LOV2-degron (20 kDa) | Blue light | 1 | Light-induced protein degradation | [65,66] |
| GLIMPSe | eLOV-TEVs-degron (27 kDa) | Blue light | 2 | Light-induced protein stabilization | [67] |
| Small-molecule-dependent photolytic peptide | SQS C-terminal peptide 371–417 (3 kDa) | YM-53601 & UV | 1 | Drug- and UV-induced protein degradation, no use of cellular degradation mechanisms | [68] |
| deGradFP | GFP (25 kDa) | NA | 2 | Protein degradation upon expression of the F-box protein, anti-GFP nanobody, no need to attach tags to POIs | [71] |
| LiPD | NA | Blue light | 2 | Light-induced protein degradation, no need to attach tags to POIs | [74] |
| DiPD | NA | Rapamycin | 2 | Drug-induced protein degradation, no need to attach tags to POIs | [74] |
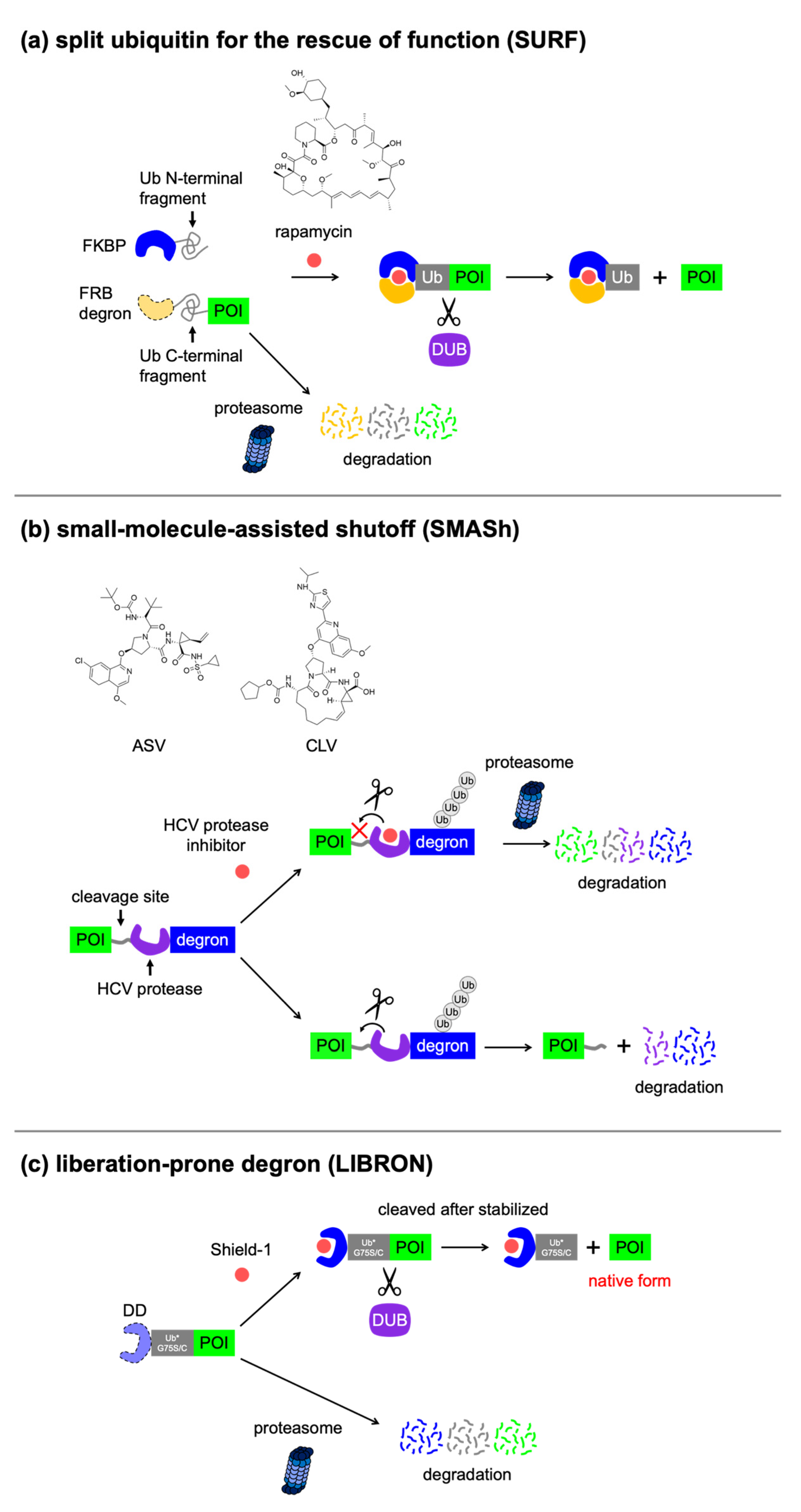
| SURF | 3 x FRB degron-Ub-C (58 kDa) | Rapamycin | 2 | Drug-induced protein stabilization, tag removal via endogenous protease activity | [75] |
| SMASh | SMASh tag (34 kDa) | HCV protease inhibitors (ASV, CLV) | 1 | Drug-induced protein degradation, tag removal via intramolecular protease activity | [76] |
| LIBRON | FKBP DD-Ub (20 kDa) DHFR DD-Ub (27 kDa) | Shield-1 (FKBP DD-Ub), TMP (DHFR DD-Ub) | 1 | Drug-induced protein stabilization, tag removal via endogenous protease activity | [77] |
4.4. Excisable Degrons
5. Applications
5.1. Drug Discovery
5.2. Gene and Cell Therapies
5.3. Research Tools
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rakhit, R.; Navarro, R.; Wandless, T.J. Chemical biology strategies for posttranslational control of protein function. Chem. Biol. 2014, 21, 1238–1252. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B. Cre/lox: One more step in the taming of the genome. Endocrine 2002, 19, 221–228. [Google Scholar] [CrossRef]
- Le, Y.; Sauer, B. Conditional gene knockout using Cre recombinase. Mol. Biotechnol. 2001, 17, 269–275. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Teng, X.; Dayhoff-Brannigan, M.; Cheng, W.C.; Gilbert, C.E.; Sing, C.N.; Diny, N.L.; Wheelan, S.J.; Dunham, M.J.; Boeke, J.D.; Pineda, F.J.; et al. Genome-wide consequences of deleting any single gene. Mol. Cell 2013, 52, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistner, A.; Gossen, M.; Zimmermann, F.; Jerecic, J.; Ullmer, C.; Lübbert, H.; Bujard, H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10933–10938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Zheng, T.; Lee, C.G.; Homer, R.J.; Elias, J.A. Tetracycline-controlled transcriptional regulation systems: Advances and application in transgenic animal modeling. Semin. Cell Dev. Biol. 2002, 13, 121–128. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Sacher, R.; Stergiou, L.; Pelkmans, L. Lessons from genetics: Interpreting complex phenotypes in RNAi screens. Curr. Opin. Cell Biol. 2008, 20, 483–489. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Ann. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef] [PubMed]
- Atilaw, Y.; Poongavanam, V.; Svensson Nilsson, C.; Nguyen, D.; Giese, A.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med. Chem. Lett. 2021, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [Green Version]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef] [Green Version]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Clift, D.; McEwan, W.A.; Labzin, L.I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 2017, 171, 1692–1706.e18. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Santos, A.F.; Mukadam, A.S.; Osswald, M.; Jacques, D.A.; Dickson, C.F.; McLaughlin, S.H.; Johnson, C.M.; Kiss, L.; Luptak, J.; et al. Target-induced clustering activates Trim-Away of pathogens and proteins. Nat. Struct. Mol. Biol. 2021, 28, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. Naming a targeting signal. Cell 1991, 64, 13–15. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Chen, L.C.; Maynard-Smith, L.A.; Ooi, A.G.; Wandless, T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 2006, 126, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Bjorklund, T.; Lundberg, C.; Kirik, D.; Wandless, T.J. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010, 17, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; Imoto, H.; Chen, L.C.; Wandless, T.J. Destabilizing domains derived from the human estrogen receptor. J. Am. Chem. Soc. 2012, 134, 3942–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, R.; Chen, L.C.; Rakhit, R.; Wandless, T.J. A Novel Destabilizing Domain Based on a Small-Molecule Dependent Fluorophore. ACS Chem. Biol. 2016, 11, 2101–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banaszynski, L.A.; Sellmyer, M.A.; Contag, C.H.; Wandless, T.J.; Thorne, S.H. Chemical control of protein stability and function in living mice. Nat. Med. 2008, 14, 1123–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.M.; Goldberg, D.E. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat. Methods 2007, 4, 1007–1009. [Google Scholar] [CrossRef]
- Herm-Gotz, A.; Agop-Nersesian, C.; Munter, S.; Grimley, J.S.; Wandless, T.J.; Frischknecht, F.; Meissner, M. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods 2007, 4, 1003–1005. [Google Scholar] [CrossRef] [Green Version]
- Cho, U.; Zimmerman, S.M.; Chen, L.C.; Owen, E.; Kim, J.V.; Kim, S.K.; Wandless, T.J. Rapid and tunable control of protein stability in Caenorhabditis elegans using a small molecule. PLoS ONE 2013, 8, e72393. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S.; Wang, J.W. A versatile genetic tool for post-translational control of gene expression in Drosophila melanogaster. eLife 2017, 6, e30327. [Google Scholar] [CrossRef] [PubMed]
- Ramadurgum, P.; Daniel, S.; Hulleman, J.D. Protocol for In Vivo Evaluation and Use of Destabilizing Domains in the Eye, Liver, and Beyond. STAR Protoc. 2020, 1, 100094. [Google Scholar] [CrossRef]
- Bonger, K.M.; Chen, L.C.; Liu, C.W.; Wandless, T.J. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011, 7, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Kubota, H. Quality control against misfolded proteins in the cytosol: A network for cell survival. J. Biochem. 2009, 146, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Chu, T.-T.; Li, Q.-Q.; Lim, Y.-J.; Qiu, T.; Ma, M.-R.; Hu, Z.-W.; Yang, X.-F.; Chen, Y.-X.; Zhao, Y.-F.; et al. Hydrophobic tagging-mediated degradation of Alzheimer’s disease related Tau. RSC Adv. 2017, 7, 40362–40366. [Google Scholar] [CrossRef] [Green Version]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef]
- Yesbolatova, A.; Saito, Y.; Kitamoto, N.; Makino-Itou, H.; Ajima, R.; Nakano, R.; Nakaoka, H.; Fukui, K.; Gamo, K.; Tominari, Y.; et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 2020, 11, 5701. [Google Scholar] [CrossRef]
- Costa, E.A.; Subramanian, K.; Nunnari, J.; Weissman, J.S. Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science 2018, 359, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Bence, M.; Jankovics, F.; Lukácsovich, T.; Erdélyi, M. Combining the auxin-inducible degradation system with CRISPR/Cas9-based genome editing for the conditional depletion of endogenous Drosophila melanogaster proteins. FEBS J. 2017, 284, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ward, J.D.; Cheng, Z.; Dernburg, A.F. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 2015, 142, 4374–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Prasanna, X.; Salo, V.T.; Vattulainen, I.; Ikonen, E. An efficient auxin-inducible degron system with low basal degradation in human cells. Nat. Methods 2019, 16, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Yamada, R.; Hagihara, S.; Iwasaki, R.; Uchida, N.; Kamura, T.; Takahashi, K.; Torii, K.U.; Fukagawa, T. A super-sensitive auxin-inducible degron system with an engineered auxin-TIR1 pair. Nucleic Acids Res. 2020, 48, e108. [Google Scholar] [CrossRef] [PubMed]
- Koduri, V.; McBrayer, S.K.; Liberzon, E.; Wang, A.C.; Briggs, K.J.; Cho, H.; Kaelin, W.G., Jr. Peptidic degron for IMiD-induced degradation of heterologous proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 2539–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S.; Shoya, Y.; Matsuoka, S.; Nishida-Fukuda, H.; Shibata, N.; Sawasaki, T. An IMiD-induced SALL4 degron system for selective degradation of target proteins. Commun. Biol. 2020, 3, 515. [Google Scholar] [CrossRef] [PubMed]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 2008, 1786, 153–177. [Google Scholar] [CrossRef]
- Renicke, C.; Schuster, D.; Usherenko, S.; Essen, L.O.; Taxis, C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 2013, 20, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonger, K.M.; Rakhit, R.; Payumo, A.Y.; Chen, J.K.; Wandless, T.J. General method for regulating protein stability with light. ACS Chem. Biol. 2014, 9, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Mondal, P.; Krishnamurthy, V.V.; Sharum, S.R.; Haack, N.; Zhou, H.; Cheng, J.; Yang, J.; Zhang, K. Repurposing Protein Degradation for Optogenetic Modulation of Protein Activities. ACS Synth. Biol. 2019, 8, 2585–2592. [Google Scholar] [CrossRef]
- Takemoto, Y.; Mao, D.; Punzalan, L.L.; Gotze, S.; Sato, S.I.; Uesugi, M. Discovery of a Small-Molecule-Dependent Photolytic Peptide. J. Am. Chem. Soc. 2020, 142, 1142–1146. [Google Scholar] [CrossRef]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Stuber, J.C.; Ernst, P.; Koch, A.; Bojar, D.; Batyuk, A.; Pluckthun, A. Design and applications of a clamp for Green Fluorescent Protein with picomolar affinity. Sci. Rep. 2017, 7, 16292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caussinus, E.; Kanca, O.; Affolter, M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2011, 19, 117–121. [Google Scholar] [CrossRef]
- Ludwicki, M.B.; Li, J.; Stephens, E.A.; Roberts, R.W.; Koide, S.; Hammond, P.T.; DeLisa, M.P. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866. [Google Scholar] [CrossRef] [Green Version]
- Daniel, K.; Icha, J.; Horenburg, C.; Muller, D.; Norden, C.; Mansfeld, J. Conditional control of fluorescent protein degradation by an auxin-dependent nanobody. Nat. Commun. 2018, 9, 3297. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Bates, J.A.; Wei, H.; Bartoschek, M.D.; Conradt, B.; Leonhardt, H. Tunable light and drug induced depletion of target proteins. Nat. Commun. 2020, 11, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, M.R.; Schwartz, E.C.; Muir, T.W. Small-molecule-mediated rescue of protein function by an inducible proteolytic shunt. Proc. Natl. Acad. Sci. USA 2007, 104, 11209–11214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Jacobs, C.L.; Huo, Y.; Yang, J.; Krumm, S.A.; Plemper, R.K.; Tsien, R.Y.; Lin, M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015, 11, 713–720. [Google Scholar] [CrossRef]
- Miyamae, Y.; Chen, L.C.; Utsugi, Y.; Farrants, H.; Wandless, T.J. A Method for Conditional Regulation of Protein Stability in Native or Near-Native Form. Cell Chem. Biol. 2020, 27, 1573–1581.e3. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharm. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef]
- Riching, K.M.; Mahan, S.; Corona, C.R.; McDougall, M.; Vasta, J.D.; Robers, M.B.; Urh, M.; Daniels, D.L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharm. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, e45457. [Google Scholar] [CrossRef]
- Qu, J.; Ren, X.; Xue, F.; He, Y.; Zhang, R.; Zheng, Y.; Huang, H.; Wang, W.; Zhang, J. Specific Knockdown of alpha-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chem. Biol. 2020, 27, 751–762.e4. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angew. Chem. Int. Ed. 2017, 56, 11530–11533. [Google Scholar] [CrossRef]
- Chang, Z.L.; Chen, Y.Y. CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol. Med. 2017, 23, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019, 19, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richman, S.A.; Wang, L.C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther. 2020, 28, 1600–1613. [Google Scholar] [CrossRef]
- Carbonneau, S.; Sharma, S.; Peng, L.; Rajan, V.; Hainzl, D.; Henault, M.; Yang, C.; Hale, J.; Shulok, J.; Tallarico, J.; et al. An IMiD-inducible degron provides reversible regulation for chimeric antigen receptor expression and activity. Cell Chem. Biol. 2021, 28, 802–812.e6. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Won, S.T.; Pentecost, M.; Bartkowski, W.; Lee, B. CRISPR/Cas9 allows efficient and complete knock-in of a destabilization domain-tagged essential protein in a human cell line, allowing rapid knockdown of protein function. PLoS ONE 2014, 9, e95101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Zhang, B.; Li, M.; Mo, F.; Mi, T.; Wu, Y.; Teng, Z.; Zhou, Q.; Li, W.; Hu, B. Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome. Nat. Commun. 2019, 10, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senturk, S.; Shirole, N.H.; Nowak, D.G.; Corbo, V.; Pal, D.; Vaughan, A.; Tuveson, D.A.; Trotman, L.C.; Kinney, J.B.; Sordella, R. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun. 2017, 8, 14370. [Google Scholar] [CrossRef]
- Sreekanth, V.; Zhou, Q.; Kokkonda, P.; Bermudez-Cabrera, H.C.; Lim, D.; Law, B.K.; Holmes, B.R.; Chaudhary, S.K.; Pergu, R.; Leger, B.S.; et al. Chemogenetic System Demonstrates That Cas9 Longevity Impacts Genome Editing Outcomes. ACS Cent. Sci. 2020, 6, 2228–2237. [Google Scholar] [CrossRef]
- Kleinjan, D.A.; Wardrope, C.; Nga Sou, S.; Rosser, S.J. Drug-tunable multidimensional synthetic gene control using inducible degron-tagged dCas9 effectors. Nat. Commun. 2017, 8, 1191. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yang, L.; Chang, T.; Kandeel, F.; Yee, J.K. A Small Molecule-Controlled Cas9 Repressible System. Mol. Ther. Nucleic Acids 2020, 19, 922–932. [Google Scholar] [CrossRef]
- Lopez Del Amo, V.; Leger, B.S.; Cox, K.J.; Gill, S.; Bishop, A.L.; Scanlon, G.D.; Walker, J.A.; Gantz, V.M.; Choudhary, A. Small-Molecule Control of Super-Mendelian Inheritance in Gene Drives. Cell Rep. 2020, 31, 107841. [Google Scholar] [CrossRef]
- Sando, R., 3rd; Baumgaertel, K.; Pieraut, S.; Torabi-Rander, N.; Wandless, T.J.; Mayford, M.; Maximov, A. Inducible control of gene expression with destabilized Cre. Nat. Methods 2013, 10, 1085–1088. [Google Scholar] [CrossRef]
- Pedone, E.; Postiglione, L.; Aulicino, F.; Rocca, D.L.; Montes-Olivas, S.; Khazim, M.; di Bernardo, D.; Pia Cosma, M.; Marucci, L. A tunable dual-input system for on-demand dynamic gene expression regulation. Nat. Commun. 2019, 10, 4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, L.Y.; Ma, H.T.; Liu, J.C.Y.; Huang, X.; Lee, N.; Poon, R.Y.C. Conditional gene inactivation by combining tetracycline-mediated transcriptional repression and auxin-inducible degron-mediated degradation. Cell Cycle 2019, 18, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef]
- Takahashi, D.; Arimoto, H. Selective autophagy as the basis of autophagy-based degraders. Cell Chem. Biol. 2021, 28, 1061–1071. [Google Scholar] [CrossRef]
- Kaiho-Soma, A.; Akizuki, Y.; Igarashi, K.; Endo, A.; Shoda, T.; Kawase, Y.; Demizu, Y.; Naito, M.; Saeki, Y.; Tanaka, K.; et al. TRIP12 promotes small-molecule-induced degradation through K29/K48-branched ubiquitin chains. Mol. Cell 2021, 81, 1411–1424.e7. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utsugi, Y.; Miyamae, Y. Strategies for Post-Translational Control of Protein Expression and Their Applications. Appl. Sci. 2021, 11, 8300. https://doi.org/10.3390/app11188300
Utsugi Y, Miyamae Y. Strategies for Post-Translational Control of Protein Expression and Their Applications. Applied Sciences. 2021; 11(18):8300. https://doi.org/10.3390/app11188300
Chicago/Turabian StyleUtsugi, Yuki, and Yusaku Miyamae. 2021. "Strategies for Post-Translational Control of Protein Expression and Their Applications" Applied Sciences 11, no. 18: 8300. https://doi.org/10.3390/app11188300
APA StyleUtsugi, Y., & Miyamae, Y. (2021). Strategies for Post-Translational Control of Protein Expression and Their Applications. Applied Sciences, 11(18), 8300. https://doi.org/10.3390/app11188300