
Accurate Prediction and Key Feature Recognition of Immunoglobulin



Abstract
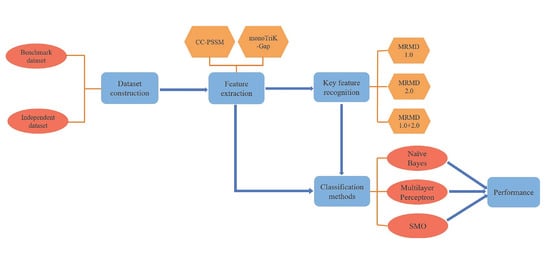
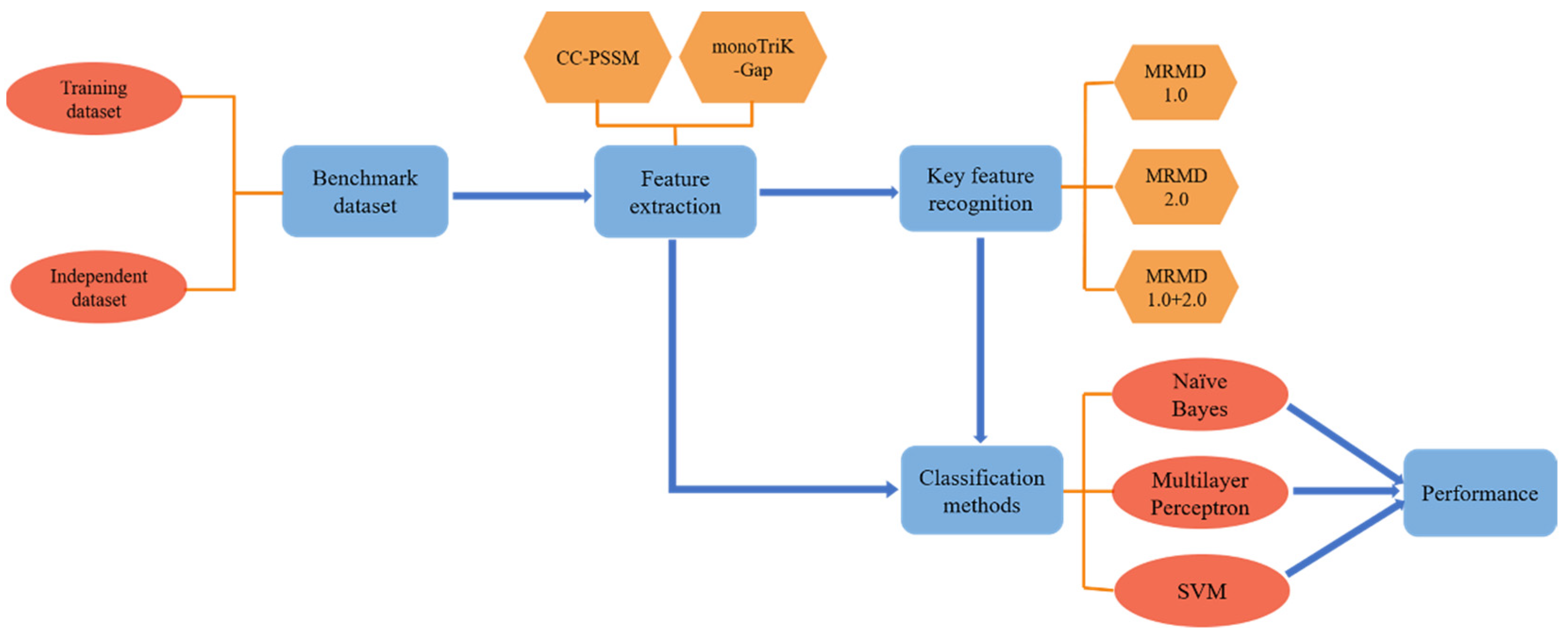
:
1. Introduction
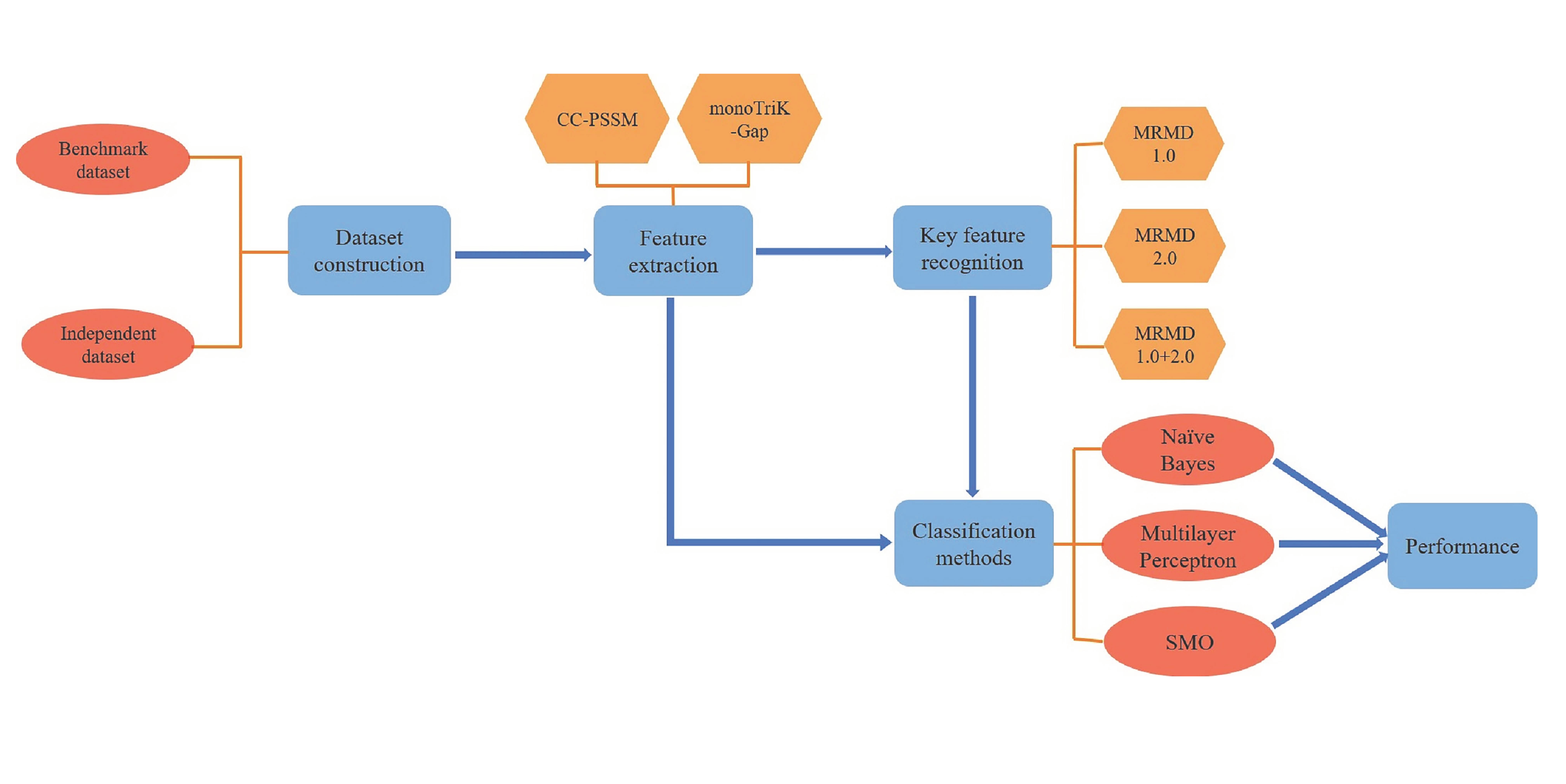
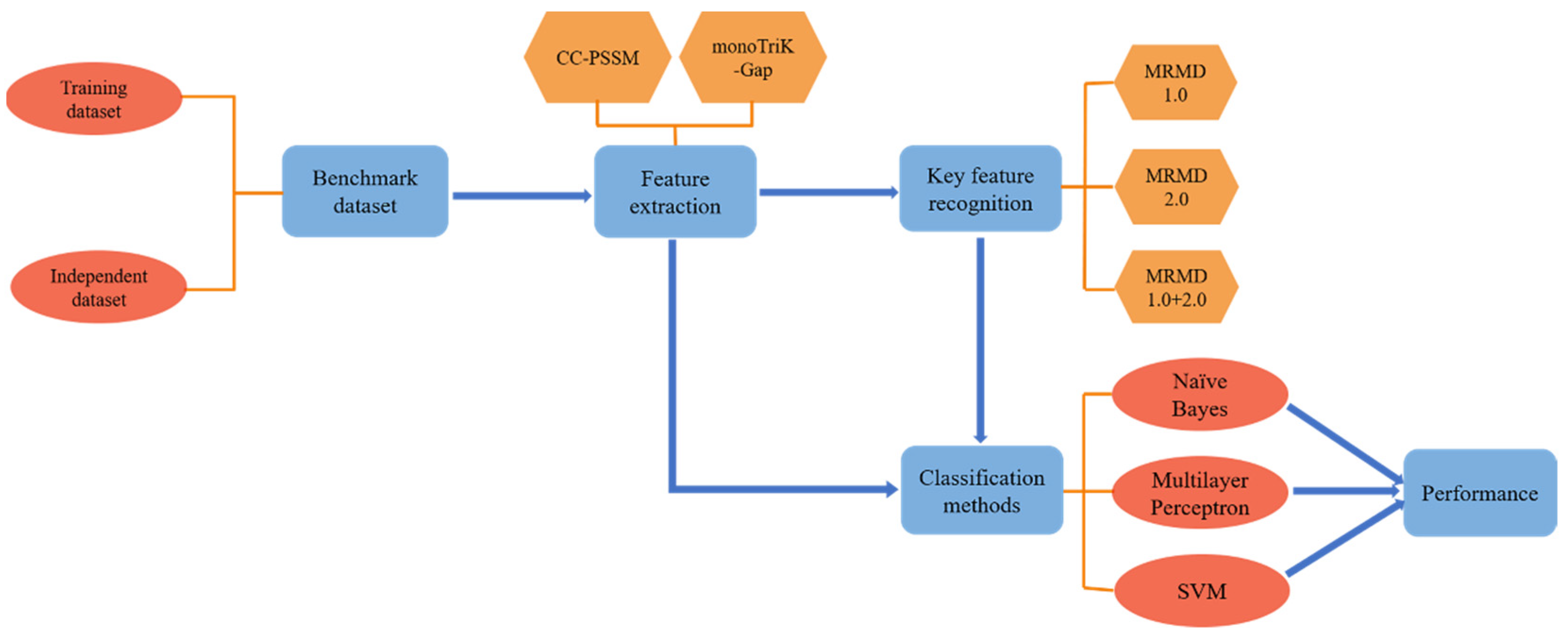
2. Materials and Methods
- Building datasets;
- CC–PSSM and monoTriKGap were selected as feature representation methods to obtain feature sets;
- MRMD1.0 and MRMD2.0 feature selection methods were selected to acquire two–dimensional key features and two–dimensional mixed key features, respectively;
- Three classifiers, Naïve Bayes, SVM, and multilayer perceptron, were selected for k–fold cross–validation to predict immunoglobulins.
2.1. Dataset Construction
2.2. Feature Extraction
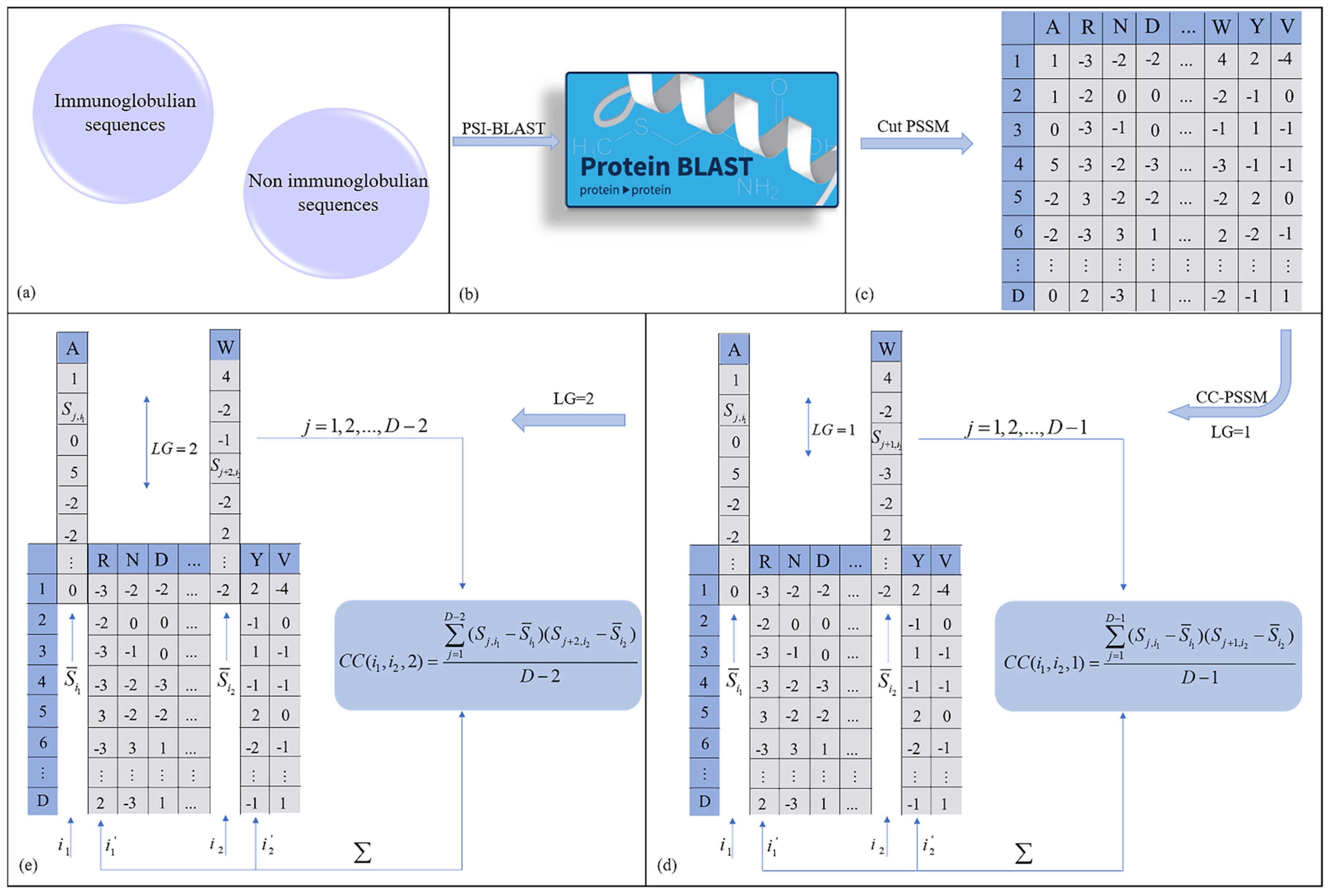
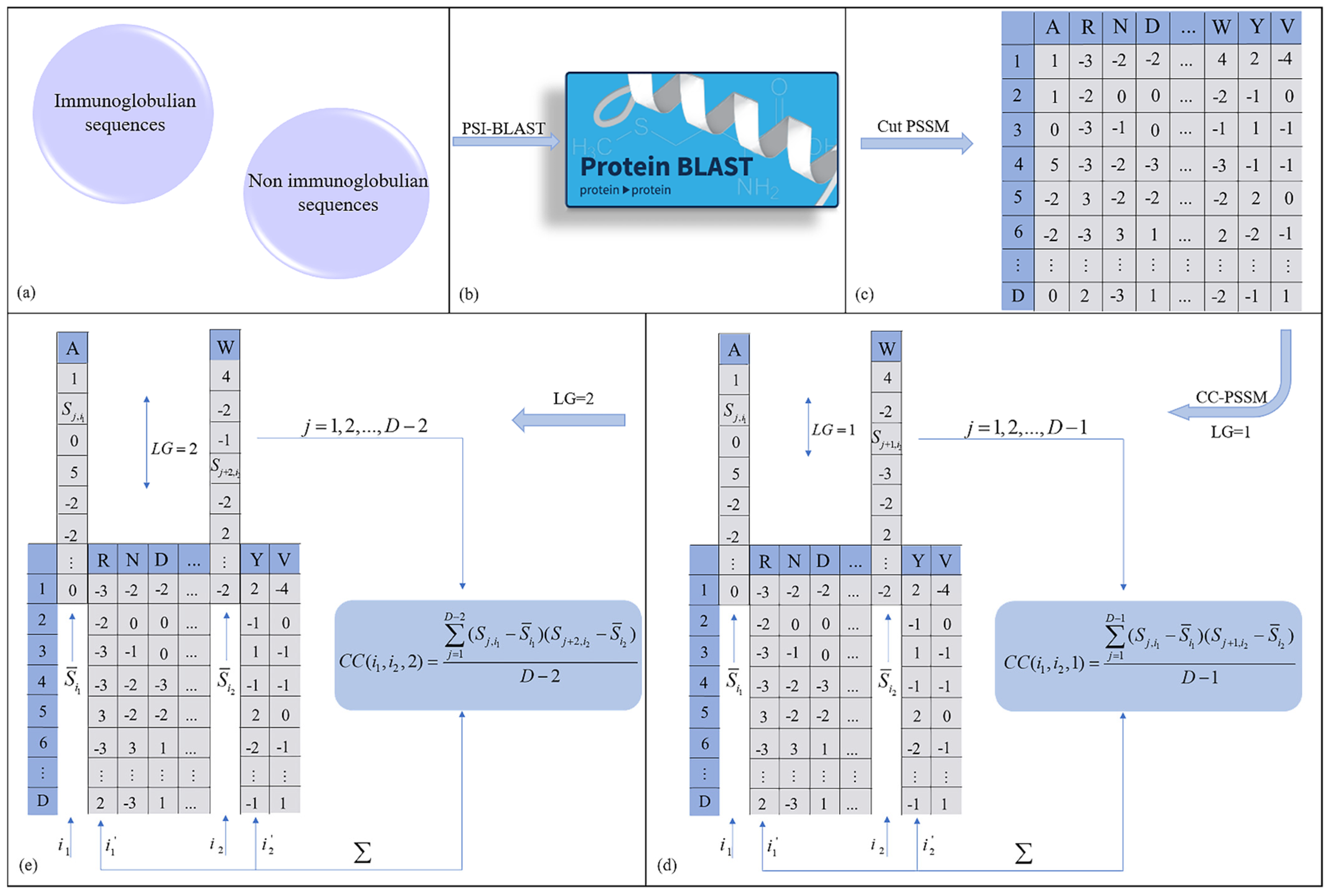
2.2.1. Profile–Based Cross Covariance (CC–PSSM)
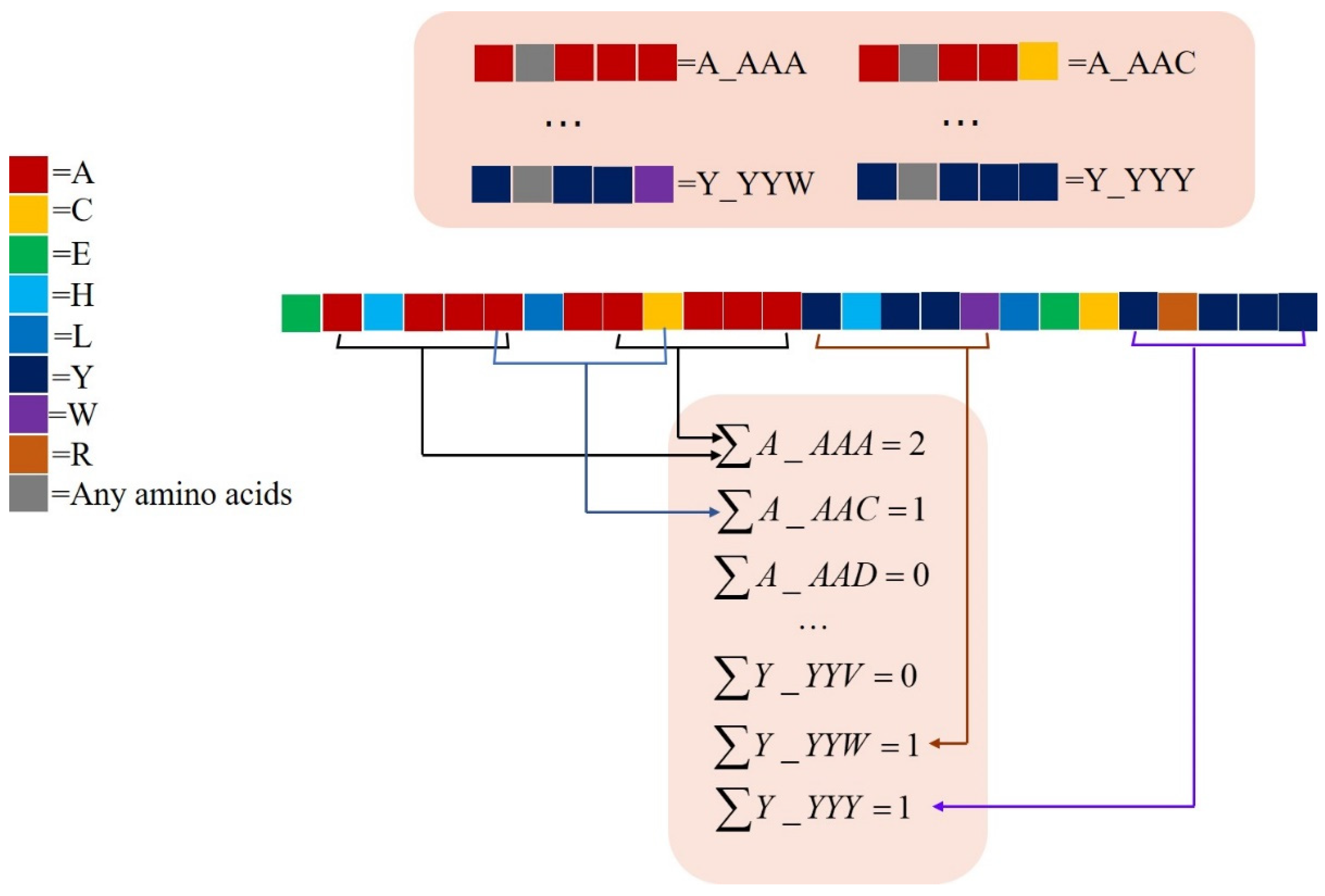
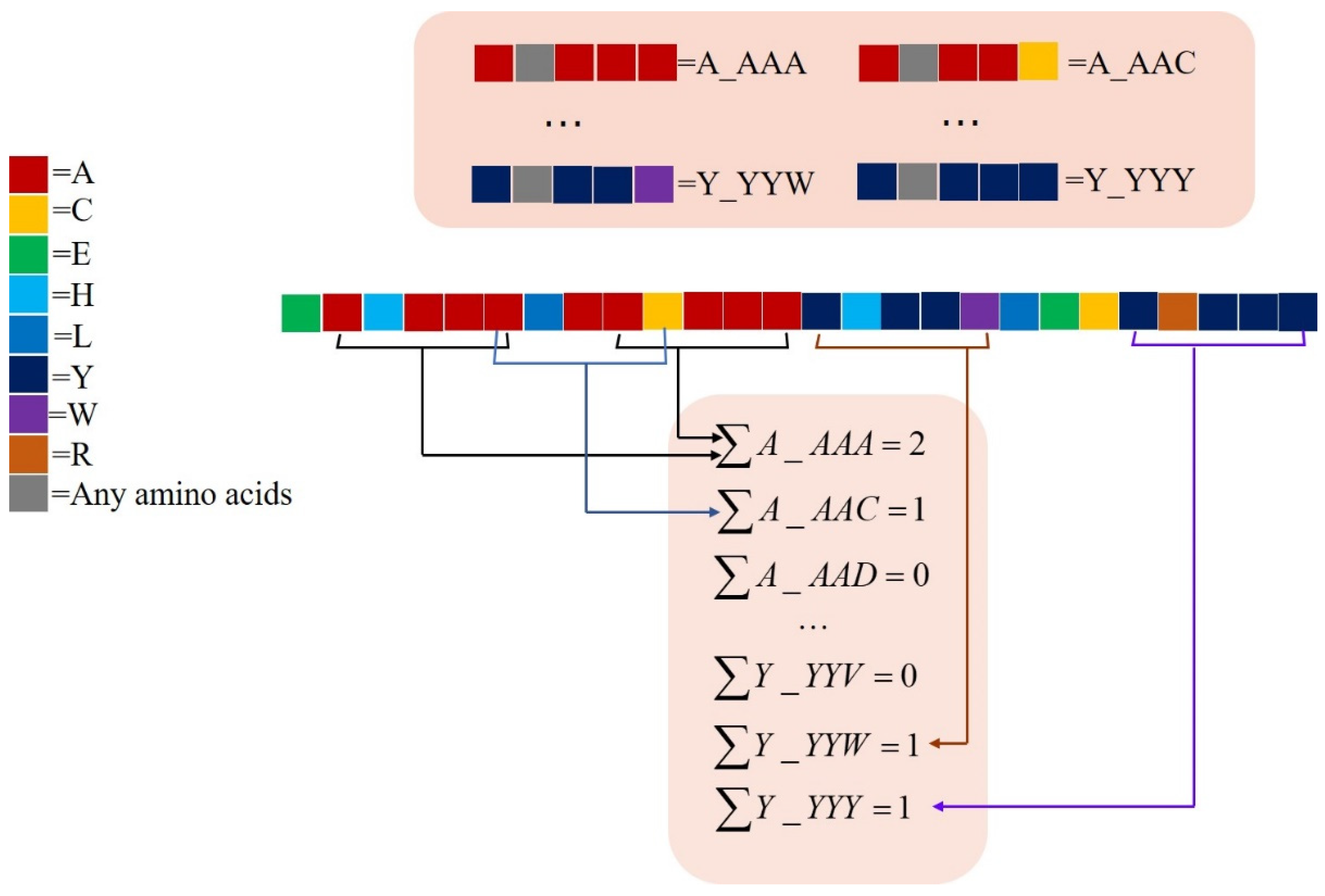
2.2.2. monoTriKGap
2.3. Classifier
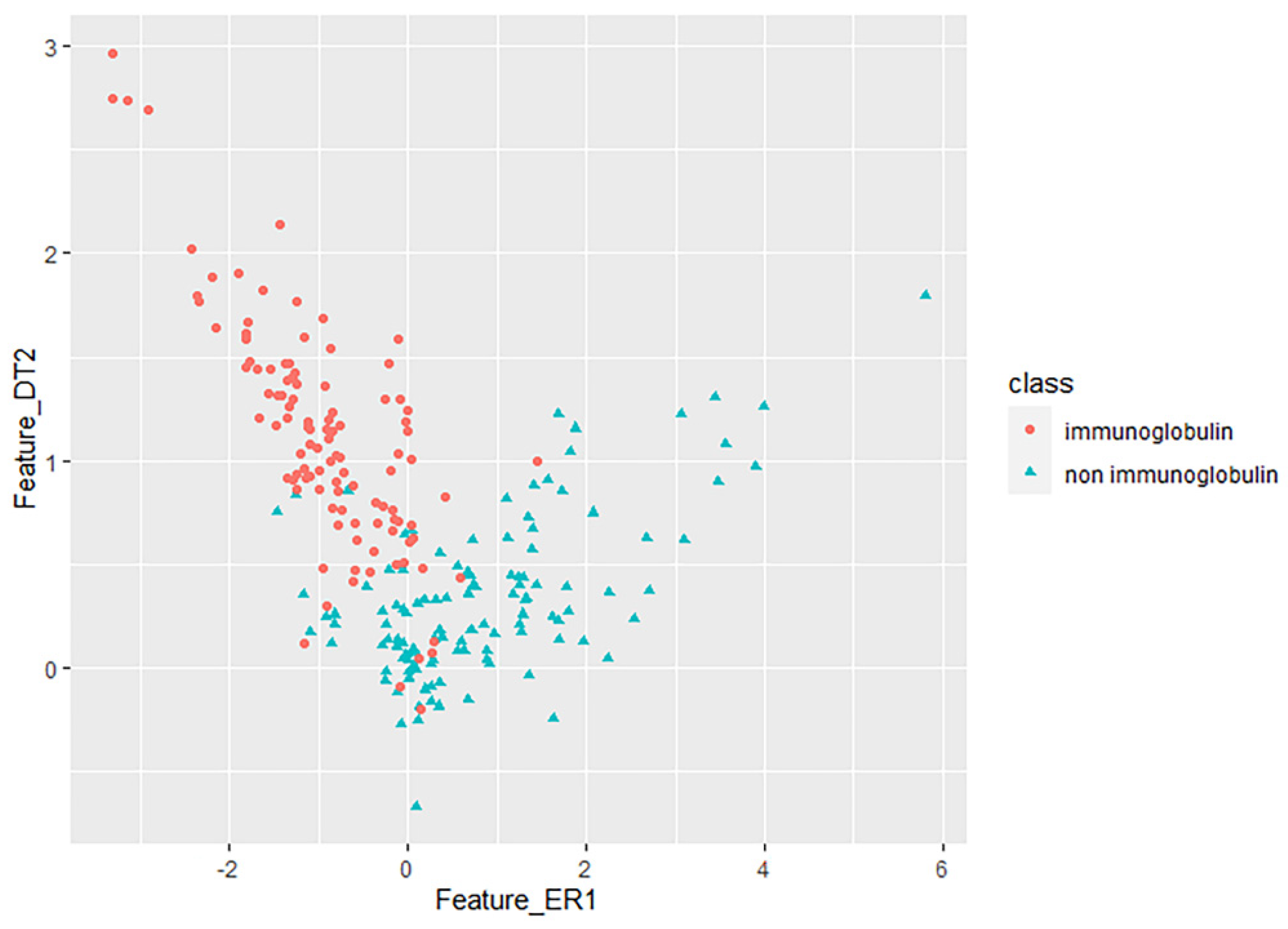
2.4. Key Feature Recognition
2.5. Performance Evaluation
3. Results and Discussion
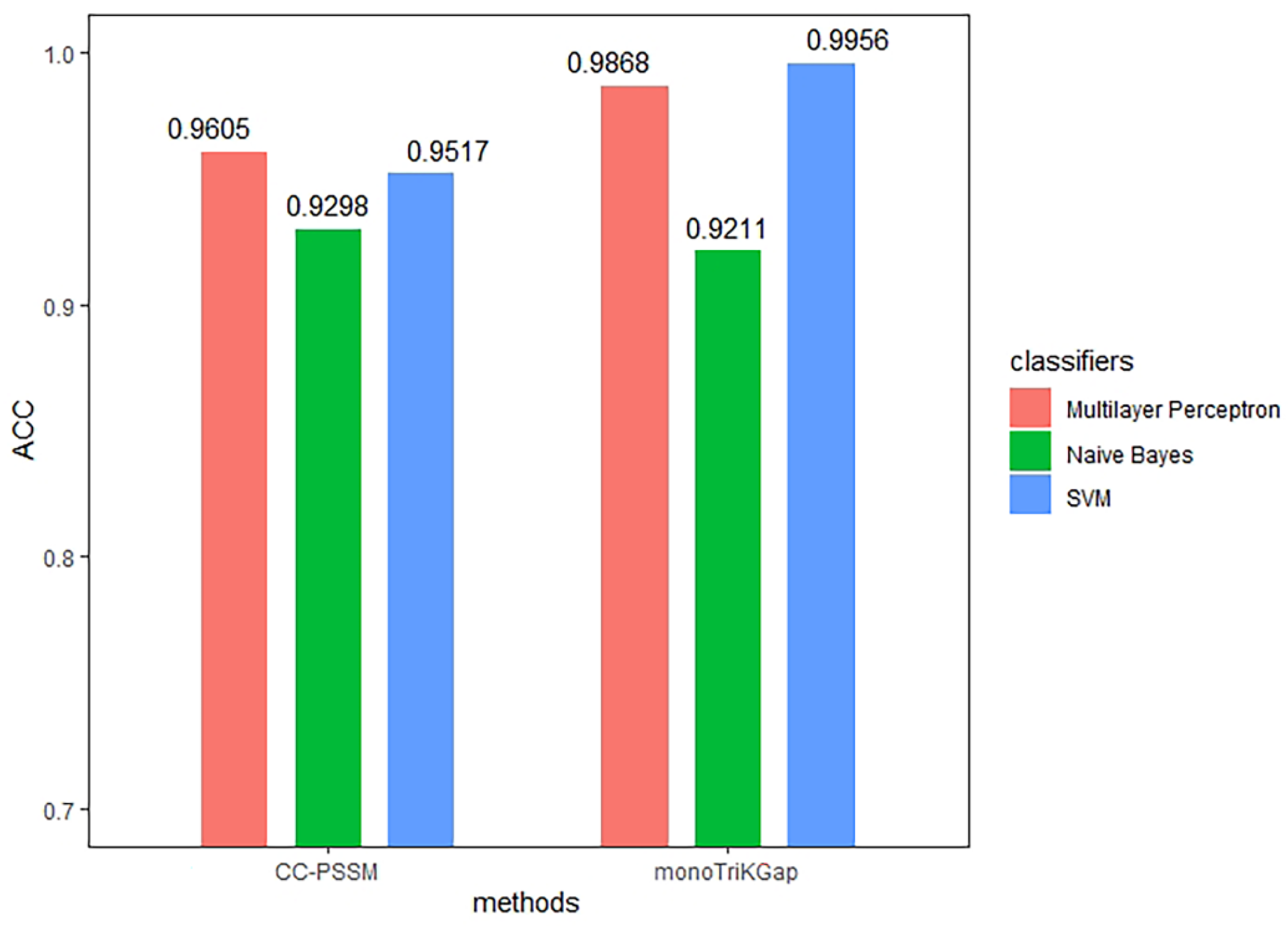
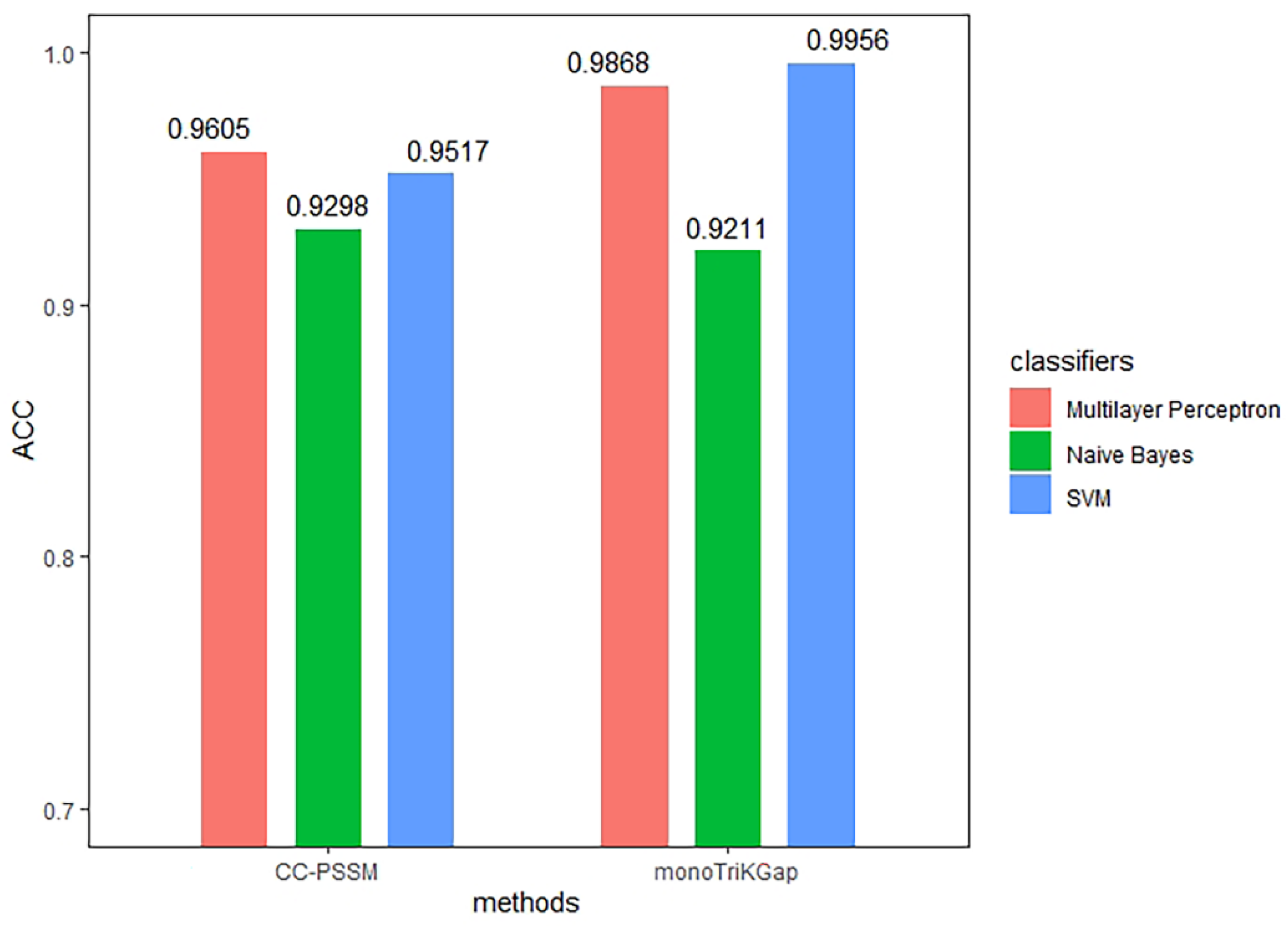
3.1. Comparison of Different Feature Extraction and Classification Methods
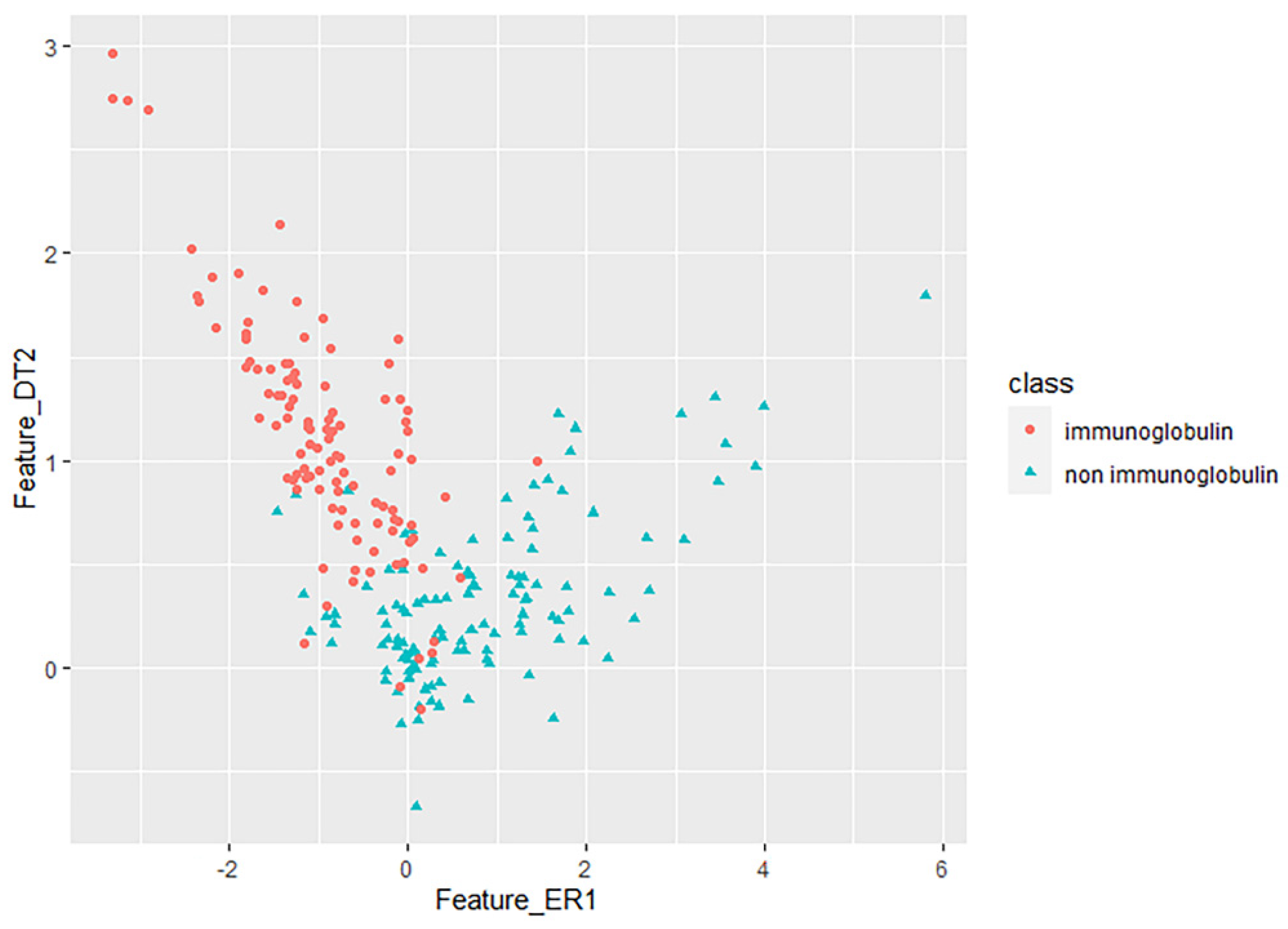
3.2. Key Feature Analysis
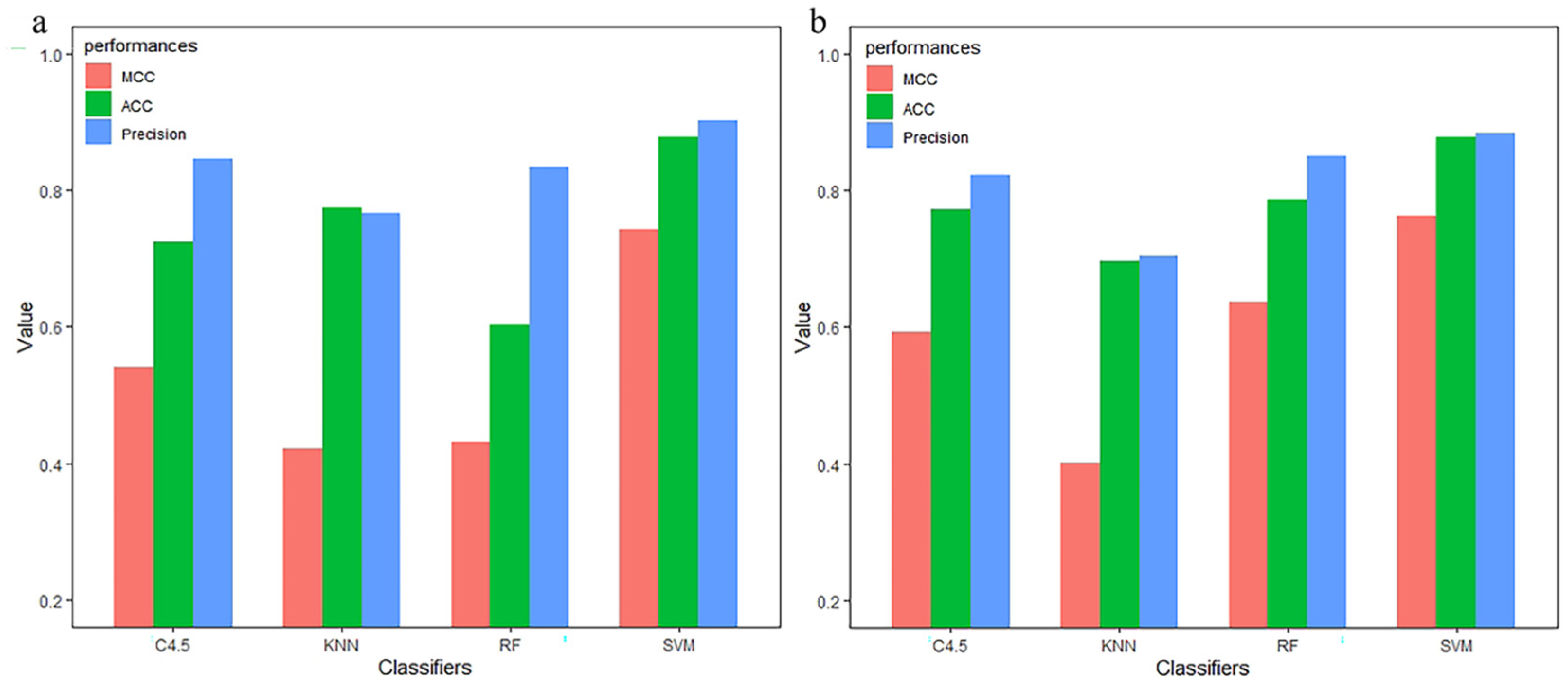
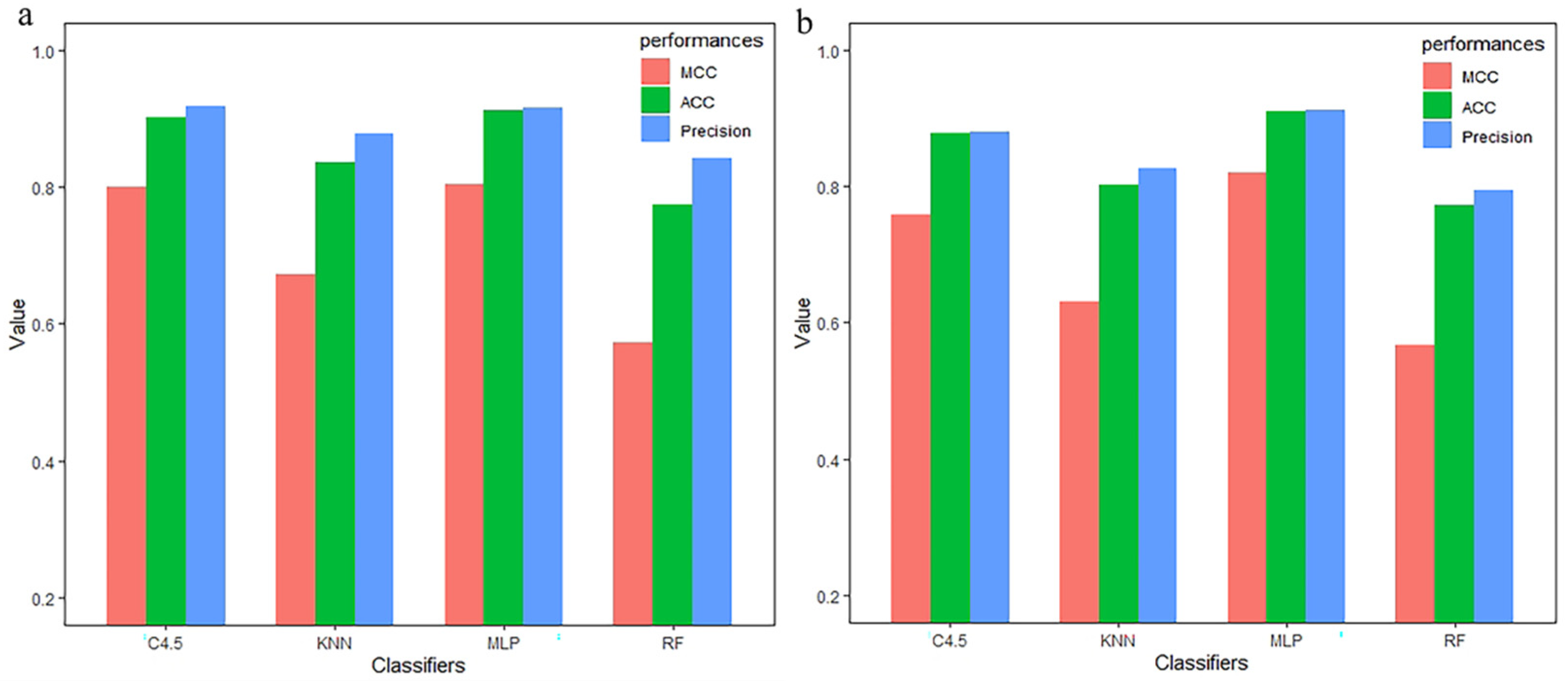
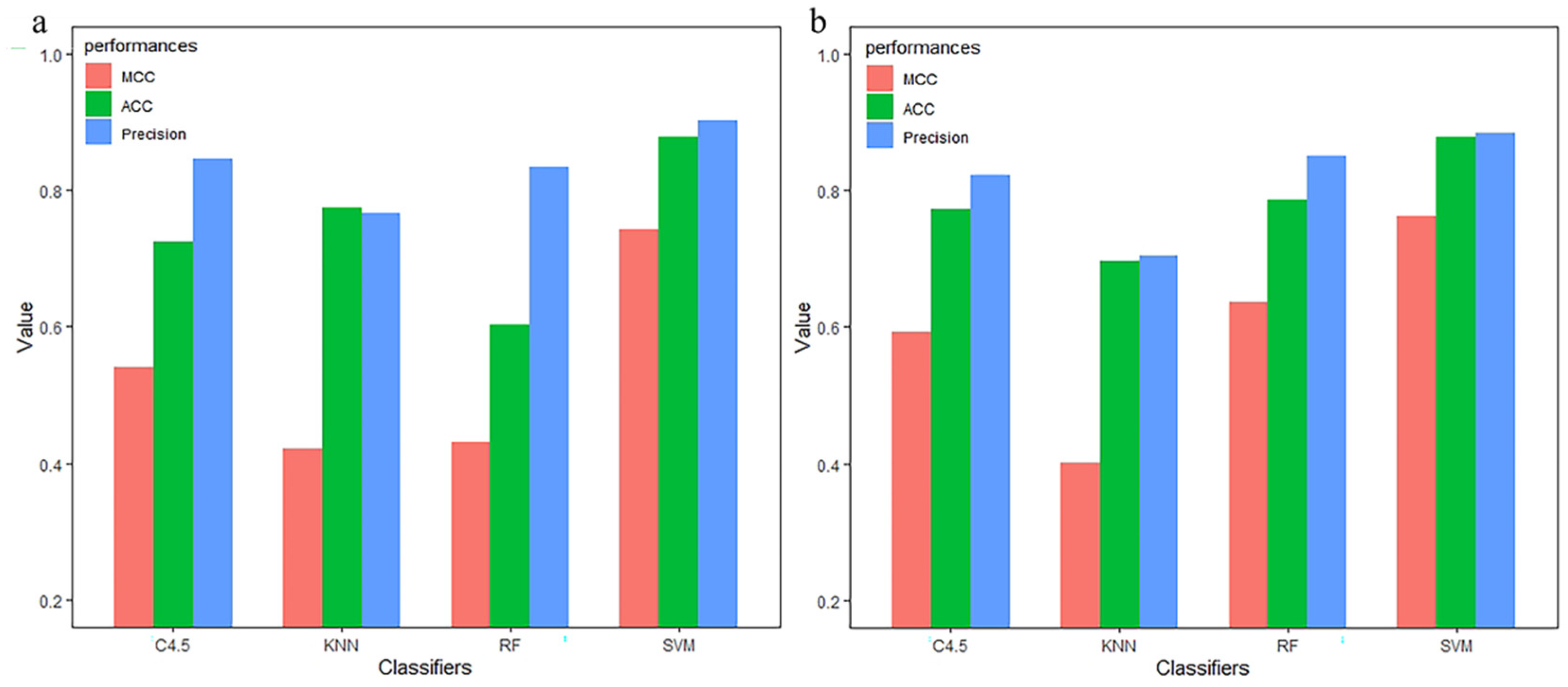
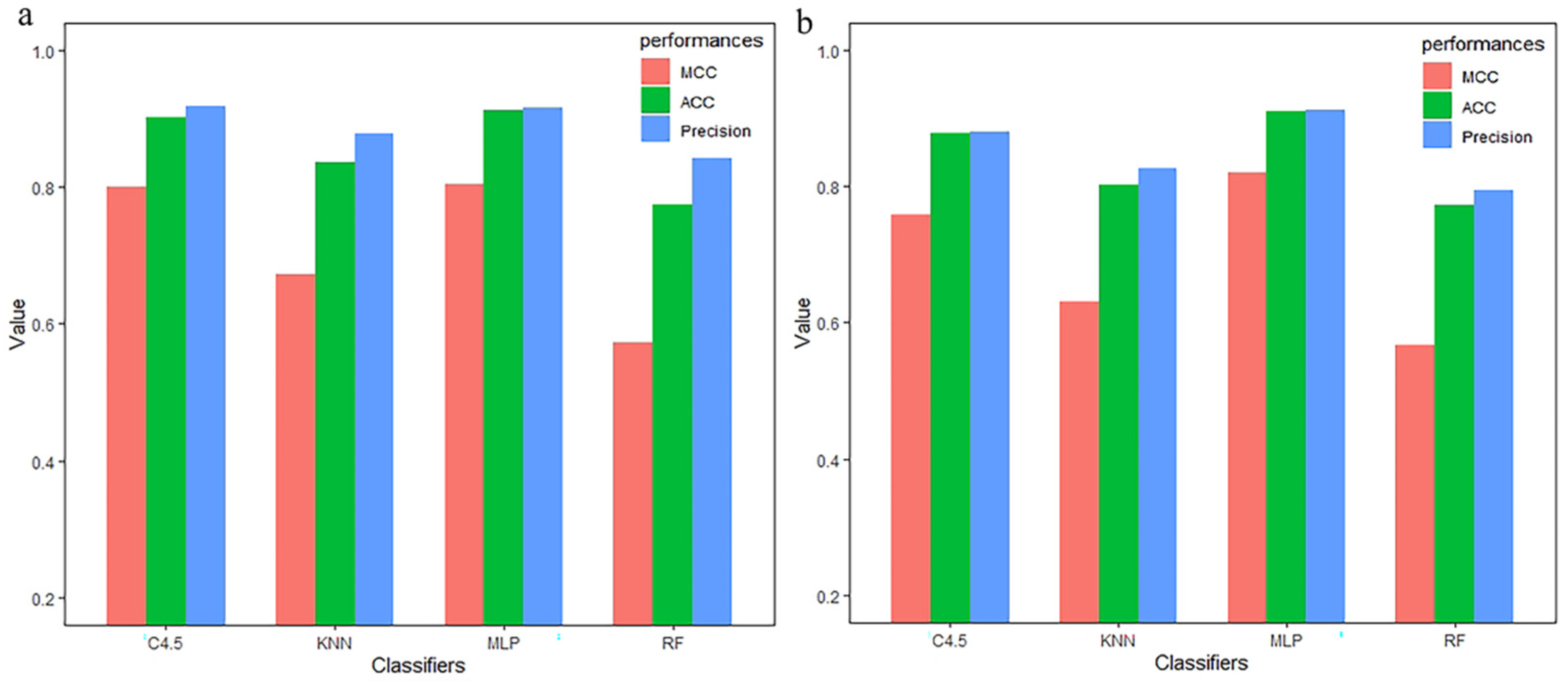
3.3. Compared with Other Classifiers
3.4. Independent Test Set Evaluation
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almaghlouth, I.; Johnson, S.R.; Pullenayegum, E.; Gladman, D.; Urowitz, M. Immunoglobulin levels in systemic lupus erythematosus: A narrative review. Lupus 2021, 30, 867–875. [Google Scholar] [CrossRef]
- Gomes, J.P.; Santos, L.; Shoenfeld, Y. Intravenous immunoglobulin (IVIG) in the vanguard therapy of Systemic Sclerosis. Clin. Immunol. 2019, 199, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Stromillo, M.L.; Vitale, A.; Lopalco, G.; Emmi, G.; Silvestri, E.; Federico, A.; Galeazzi, M.; Iannone, F.; De Stefano, N. Efficacy and Safety of Intravenous Immunoglobulin Treatment in Refractory Behcet’s Disease with Different Organ Involvement: A Case Series. Isr. Med. Assoc. J. 2016, 18, 238–242. [Google Scholar] [PubMed]
- Tenti, S.; Fabbroni, M.; Mancini, V.; Russo, F.; Galeazzi, M.; Fioravanti, A. Intravenous Immunoglobulins as a new opportunity to treat discoid lupus erythematosus: A case report and review of the literature. Autoimmun. Rev. 2018, 17, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, M.; Yang, Y.; Xu, F.; Zhang, X.; Xie, F.; Gao, L.; Li, X. Predicting therapeutic drugs for hepatocellular carcinoma based on tissue–specific pathways. PLoS Comput. Biol. 2021, 17, e1008696. [Google Scholar] [CrossRef] [PubMed]
- Marcatili, P.; Olimpieri, P.P.; Chailyan, A.; Tramontano, A. Antibody structural modeling with prediction of immunoglobulin structure (PIGS). Nat. Protoc. 2014, 9, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, R.; Liu, K.; Li, L.; Zai, X.; Chi, X.; Fu, L.; Xu, J.; Chen, W. Identification of antigen–specific human monoclonal antibodies using high–throughput sequencing of the antibody repertoire. Biochem. Biophys. Res. Commun. 2016, 473, 23–28. [Google Scholar] [CrossRef]
- Salvo, P.; Vivaldi, F.M.; Bonini, A.; Biagini, D.; Bellagambi, F.G.; Miliani, F.M.; Francesco, F.D.; Lomonaco, T. Biosensors for Detecting Lymphocytes and Immunoglobulins. Biosensors 2020, 10, 155. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network–based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug–side effect association via multiple information integration with centered kernel alignment. Neurocomputing 2019, 325, 211–224. [Google Scholar] [CrossRef]
- Yu, L.; Zhou, D.; Gao, L.; Zha, Y. Prediction of drug response in multilayer networks based on fusion of multiomics data. Methods 2020. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred–FL: A sequence–based predictor using effective feature representation to improve the prediction of anti–cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Zhu, X.J.; Feng, C.Q.; Lai, H.Y.; Chen, W.; Lin, H. Predicting protein structural classes for low–similarity sequences by evaluating different features. Knowl. Based Syst. 2019, 163, 787–793. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone–binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot–PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Cai, L.; Zeng, X.; Zou, Q. StackCPPred: A stacking and pairwise energy content–based prediction of cell–penetrating peptides and their uptake efficiency. Bioinformatics 2020, 36, 3028–3034. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, X.; Zhang, H. BioSeq–Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef]
- Zhai, Y.; Chen, Y.; Teng, Z.; Zhao, Y. Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein–Protein Interactions. Front. Cell Dev. Biol. 2020, 8, 591487. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein cellular attributes using pseudo–amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wang, L.; Fu, X.; Xia, C.; Zeng, X.; Zou, Q. ITP–Pred: An interpretable method for predicting, therapeutic peptides with fused features low–dimension representation. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Tang, Y.-J.; Pang, Y.-H.; Liu, B. IDP–Seq2Seq: Identification of Intrinsically Disordered Regions based on Sequence to Sequence Learning. Bioinformaitcs 2020, 36, 5177–5186. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.X.; Li, S.H.; Zhang, Z.M.; Chen, C.X.; Chen, W.; Tang, H.; Lin, H. Identification of hormone binding proteins based on machine learning methods. Math. Biosci. Eng. 2019, 16, 2466–2480. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, J.; Guo, F. Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou’s general PseAAC. J. Theor. Biol. 2019, 462, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc–Hum: Using the accumulation–label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol. Biosyst. 2012, 8, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, K.; Huang, D.S.; Chou, K.C. iEnhancer–EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Liu, B. ProtFold–DFG: Protein fold recognition by combining Directed Fusion Graph and PageRank algorithm. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chen, H.-D.; Zulfiqar, H.; Yuan, S.-S.; Huang, Q.-L.; Zhang, Z.-Y.; Deng, K.-J. iBLP: An XGBoost–Based Predictor for Identifying Bioluminescent Proteins. Comput. Math. Methods Med. 2021, 2021, 6664362. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Li, Y.; Chen, Y.; Li, G.; Yan, Z.; Yang, L. PseKRAAC: A flexible web server for generating pseudo K–tuple reduced amino acids composition. Bioinformatics 2017, 33, 122–124. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef]
- Dong, Q.; Zhou, S.; Guan, J. A new taxonomy–based protein fold recognition approach based on autocross–covariance transformation. Bioinformatics 2009, 25, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Muhammod, R.; Ahmed, S.; Md Farid, D.; Shatabda, S.; Sharma, A.; Dehzangi, A. PyFeat: A Python–based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics 2019, 35, 3831–3833. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug–target interactions via multiple information integration. Inf. Sci. 2017, 418, 546–560. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss–Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD–HIT: Accelerated for clustering the next–generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Su, W.; Wang, J.S.; Yang, Y.H.; Yang, H.; Lin, H. Predicting Preference of Transcription Factors for Methylated DNA Using Sequence Information. Mol. Ther. Nucleic Acids 2020, 22, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, Y.; Tang, J.; Guo, F. Identification of membrane protein types via multivariate information fusion with Hilbert–Schmidt Independence Criterion. Neurocomputing 2019, 383, 257–269. [Google Scholar] [CrossRef]
- Wei, L.; He, W.; Malik, A.; Su, R.; Cui, L.; Manavalan, B. Computational prediction and interpretation of cell–specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI–BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Z.; Pu, L.; Tang, J.; Guo, F. AIEpred: An ensemble predictive model of classifier chain to identify anti–inflammatory peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying Sigma70 Promoters with Novel Pseudo Nucleotide Composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Ao, C.; Jin, S.; Ding, H.; Zou, Q.; Yu, L. Application and Development of Artificial Intelligence and Intelligent Disease Diagnosis. Curr. Pharm. Design 2020, 26, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, W.; Dao, F.Y.; Lv, H.; Ding, H.; Chen, W.; Lin, H. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 2020, 21, 1568–1580. [Google Scholar] [CrossRef]
- Wei, L.; Chen, H.; Su, R. M6APred–EL: A Sequence–Based Predictor for Identifying N6–methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [Green Version]
- Liu, B. BioSeq–Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2019, 20, 1280–1294. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wan, S.; Guo, J.; Wong, K.K. A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 2017, 83, 82–90. [Google Scholar] [CrossRef]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS ONE 2017, 12, e0177678. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug–target interactions via fuzzy bipartite local model. Neural Comput. Appl. 2020, 32, 1–17. [Google Scholar] [CrossRef]
- Sun, H. A naive bayes classifier for prediction of multidrug resistance reversal activity on the basis of atom typing. J. Med. Chem. 2005, 48, 4031–4039. [Google Scholar] [CrossRef] [PubMed]
- Yongchuan, T.; Wuming, P.; Haiming, L.; Yang, X. Fuzzy Naive Bayes classifier based on fuzzy clustering. In Proceedings of the IEEE International Conference on Systems, Man and Cybernetics, Yasmine Hammamet, Tunisia, 6–9 October 2002; Volume 5, p. 6. [Google Scholar]
- Keerthi, S.S.; Shevade, S.K.; Bhattacharyya, C.; Murthy, K.R.K. Improvements to Platt’s SMO Algorithm for SVM Classifier Design. Neural Comput. 2001, 13, 637–649. [Google Scholar] [CrossRef]
- Platt, J.C. Fast training of support vector machines using sequential minimal optimization. In Advances in Kernel Methods: Support Vector Learning; MIT Press: Cambridge, MA, USA, 1999; pp. 185–208. [Google Scholar]
- Zhang, Y.-H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.-D. Detecting the multiomics signatures of factor–specific inflammatory effects on airway smooth muscles. Front. Genet. 2021, 11, 599970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Li, H.; Zeng, T.; Chen, L.; Li, Z.; Huang, T.; Cai, Y.-D. Identifying transcriptomic signatures and rules for SARS–CoV–2 infection. Front. Cell Dev. Biol. 2021, 8, 627302. [Google Scholar] [CrossRef]
- Su, R.; Wu, H.; Bo, X.; Liu, X.; Wei, L. Developing a Multi–Dose Computational Model for Drug–Induced Hepatotoxicity Prediction Based on Toxicogenomics Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Pan, X.; Li, H.; Gardiner, A.; Sargent, I.; Hare, J.; Atkinson, P.M. A hybrid MLP–CNN classifier for very fine resolution remotely sensed image classification. ISPRS J. Photogramm. Remote Sens. 2018, 140, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Shida, H.; Fei, G.; Quan, Z.; HuiDing. MRMD2.0: A Python Tool for Machine Learning with Feature Ranking and Reduction. Curr. Bioinform. 2020, 15, 1213–1221. [Google Scholar] [CrossRef]
- Tao, Z.; Li, Y.; Teng, Z.; Zhao, Y. A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 2020, 8926750. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Zeng, X.; Wei, L.; Liu, X. Identifying enhancer–promoter interactions with neural network based on pre–trained DNA vectors and attention mechanism. Bioinformatics 2020, 36, 1037–1043. [Google Scholar] [CrossRef]
- Su, R.; Hu, J.; Zou, Q.; Manavalan, B.; Wei, L. Empirical comparison and analysis of web–based cell–penetrating peptide prediction tools. Brief. Bioinform. 2020, 21, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Liu, X.; Xiao, G.; Wei, L. Meta–GDBP: A high–level stacked regression model to improve anticancer drug response prediction. Brief. Bioinform. 2020, 21, 996–1005. [Google Scholar] [CrossRef]
- Hong, Q.; Yan, R.; Wang, C.; Sun, J. Memristive Circuit Implementation of Biological Nonassociative Learning Mechanism and Its Applications. IEEE Trans. Biomed. Circuits Syst. 2020, 14, 1036–1050. [Google Scholar] [CrossRef] [PubMed]
- Accurate prediction of potential druggable proteins based on genetic algorithm and Bagging–SVM ensemble classifier. Artif. Intell. Med. 2019, 98, 35–47. [CrossRef]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep–Resp–Forest: A deep forest model to predict anti–cancer drug response. Methods 2019, 166, 91–102. [Google Scholar] [CrossRef]
- Shao, J.; Yan, K.; Liu, B. FoldRec–C2C: Protein fold recognition by combining cluster–to–cluster model and protein similarity network. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of Drug–Target Interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion. Knowl. Based Syst. 2020, 204, 106254. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, G.; Jin, S.; Yu, L.; Wang, Y. Predicting human microRNA–disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Zeng, J.; Chen, J.; Su, R.; Guo, F. Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 2017, 83, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, J.; Ding, Y.; Guo, F. Exploring associations of non–coding RNAs in human diseases via three–matrix factorization with hypergraph–regular terms on center kernel alignment. Brief. Bioinform. 2021. [Google Scholar] [CrossRef]
- MwanjeleMwagha, S.; Muthoni, M.; Ochieng, P. Comparison of Nearest Neighbor (ibk), Regression by Discretization and Isotonic Regression Classification Algorithms for Precipitation Classes Prediction. Int. J. Comput. Appl. 2014, 96, 44. [Google Scholar] [CrossRef]
- Aljawarneh, S.; Yassein, M.B.; Aljundi, M. An enhanced J48 classification algorithm for the anomaly intrusion detection systems. Clust. Comput. 2019, 22, 10549–10565. [Google Scholar] [CrossRef]
- Rodriguez-Galiano, V.F.; Ghimire, B.; Rogan, J.; Chica-Olmo, M.; Rigol-Sanchez, J.P. An assessment of the effectiveness of a random forest classifier for land–cover classification. ISPRS J. Photogramm. Remote Sens. 2012, 67, 93–104. [Google Scholar] [CrossRef]
- Cheng, L.; Hu, Y.; Sun, J.; Zhou, M.; Jiang, Q. DincRNA: A comprehensive web–based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 2018, 34, 1953–1956. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.D. Determining protein–protein functional associations by functional rules based on gene ontology and KEGG pathway. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2021, 1869, 140621. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Length (Amino Acids) | Immunoglobulin Dataset | Non Immunoglobulin Dataset | ||||
|---|---|---|---|---|---|---|
| Human | Mouse | Rat | Human | Mouse | Rat | |
| <400 | 26 | 11 | 3 | 16 | 31 | 3 |
| 400–700 | 22 | 12 | 2 | 8 | 24 | 2 |
| >700 | 20 | 10 | 3 | 5 | 28 | 2 |
| Method | Classifier | TPR | FPR | Precision | MCC | auROC | ACC |
|---|---|---|---|---|---|---|---|
| CC–PSSM | NB | 0.930 | 0.072 | 0.930 | 0.860 | 0.951 | 0.9298 |
| MLP | 0.961 | 0.041 | 0.961 | 0.921 | 0.994 | 0.9605 | |
| SVM | 0.952 | 0.050 | 0.952 | 0.904 | 0.951 | 0.9517 | |
| monoTriKGap | NB | 0.921 | 0.081 | 0.921 | 0.842 | 0.951 | 0.9211 |
| MLP | 0.987 | 0.013 | 0.987 | 0.974 | 0.997 | 0.9868 | |
| SVM | 0.996 | 0.004 | 0.996 | 0.991 | 0.996 | 0.9956 | |
| Tang et al. [29] | SVM | 0.963 | 0.025 | \ | \ | 0.994 | 0.9690 |
| Kernel Function | TPR | FPR | Precision | MCC | auROC | ACC |
|---|---|---|---|---|---|---|
| liner kernel | 0.996 | 0.004 | 0.996 | 0.991 | 0.996 | 0.9956 |
| polynomial kernel | 0.864 | 0.138 | 0.864 | 0.728 | 0.863 | 0.8640 |
| RBF | 0.746 | 0.277 | 0.821 | 0.554 | 0.734 | 0.7456 |
| Method | Selection | Classifier | TPR | FPR | Precision | MCC | auROC | ACC |
|---|---|---|---|---|---|---|---|---|
| MRMD1.0 | NB | 0.890 | 0.113 | 0.892 | 0.781 | 0.950 | 0.8904 | |
| MLP | 0.904 | 0.101 | 0.908 | 0.810 | 0.946 | 0.9035 | ||
| SVM | 0.868 | 0.139 | 0.877 | 0.744 | 0.865 | 0.8684 | ||
| CC–PSSM | MRMD2.0 | NB | 0.846 | 0.158 | 0.849 | 0.694 | 0.875 | 0.8465 |
| MLP | 0.825 | 0.177 | 0.825 | 0.648 | 0.877 | 0.8246 | ||
| SVM | 0.803 | 0.202 | 0.804 | 0.605 | 0.801 | 0.8026 | ||
| MRMD 1.0+2.0 | NB | 0.886 | 0.121 | 0.895 | 0.780 | 0.955 | 0.8859 | |
| MLP | 0.921 | 0.081 | 0.921 | 0.842 | 0.934 | 0.9211 | ||
| SVM | 0.895 | 0.112 | 0.902 | 0.796 | 0.891 | 0.8947 | ||
| MRMD1.0 | NB | 0.535 | 0.439 | 0.576 | 0.119 | 0.563 | 0.5351 | |
| MLP | 0.526 | 0.461 | 0.537 | 0.068 | 0.505 | 0.5263 | ||
| SVM | 0.531 | 0.509 | 0.562 | 0.052 | 0.511 | 0.5307 | ||
| MonoTriKGap | MRMD2.0 | NB | 0.491 | 0.500 | 0.496 | –0.009 | 0.514 | 0.4804 |
| MLP | 0.491 | 0.499 | 0.497 | –0.008 | 0.501 | 0.4912 | ||
| SVM | 0.522 | 0.522 | 0.522 | 0.000 | 0.500 | 0.5219 | ||
| MRMD 1.0+2.0 | NB | 0.539 | 0.442 | 0.561 | 0.108 | 0.572 | 0.5395 | |
| MLP | 0.522 | 0.454 | 0.550 | 0.081 | 0.514 | 0.5219 | ||
| SVM | 0.522 | 0.522 | 0.522 | 0.000 | 0.500 | 0.5219 |
| Method | Classifier | TPR | FPR | Precision | MCC | auROC | ACC |
|---|---|---|---|---|---|---|---|
| monoTriKGap | SVM | 0.996 | 0.004 | 0.996 | 0.991 | 0.996 | 0.9956 |
| KNN | 0.732 | 0.288 | 0.787 | 0.510 | 0.831 | 0.7325 | |
| C4.5 | 0.833 | 0.169 | 0.833 | 0.666 | 0.838 | 0.8333 | |
| RF | 0.969 | 0.034 | 0.971 | 0.940 | 0.988 | 0.9693 |
| Method | Selection | Classifier | TPR | FPR | Precision | MCC | auROC | ACC |
|---|---|---|---|---|---|---|---|---|
| CC–PSSM | MRMD 1.0 + 2.0 | MLP | 0.921 | 0.081 | 0.921 | 0.842 | 0.934 | 0.9211 |
| KNN | 0.895 | 0.106 | 0.895 | 0.789 | 0.928 | 0.8947 | ||
| C4.5 | 0.904 | 0.098 | 0.904 | 0.807 | 0.897 | 0.9035 | ||
| RF | 0.886 | 0.114 | 0.886 | 0.771 | 0.930 | 0.8859 |
| Training Dataset | Independent Dataset | |||||
|---|---|---|---|---|---|---|
| Composition | Positive | Negative | Composition | Positive | Negative | |
| Group1 | human, rat | 76 | 36 | mouse | 33 | 83 |
| Group2 | human, rat | 76 | 36 | mouse | 33 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Liao, B.; Peng, D.; Zou, Q. Accurate Prediction and Key Feature Recognition of Immunoglobulin. Appl. Sci. 2021, 11, 6894. https://doi.org/10.3390/app11156894
Gong Y, Liao B, Peng D, Zou Q. Accurate Prediction and Key Feature Recognition of Immunoglobulin. Applied Sciences. 2021; 11(15):6894. https://doi.org/10.3390/app11156894
Chicago/Turabian StyleGong, Yuxin, Bo Liao, Dejun Peng, and Quan Zou. 2021. "Accurate Prediction and Key Feature Recognition of Immunoglobulin" Applied Sciences 11, no. 15: 6894. https://doi.org/10.3390/app11156894
APA StyleGong, Y., Liao, B., Peng, D., & Zou, Q. (2021). Accurate Prediction and Key Feature Recognition of Immunoglobulin. Applied Sciences, 11(15), 6894. https://doi.org/10.3390/app11156894