Recovery of Li and Co from LiCoO2 via Hydrometallurgical–Electrodialytic Treatment

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Extraction Analysis
2.2. Solid Surface Characterization
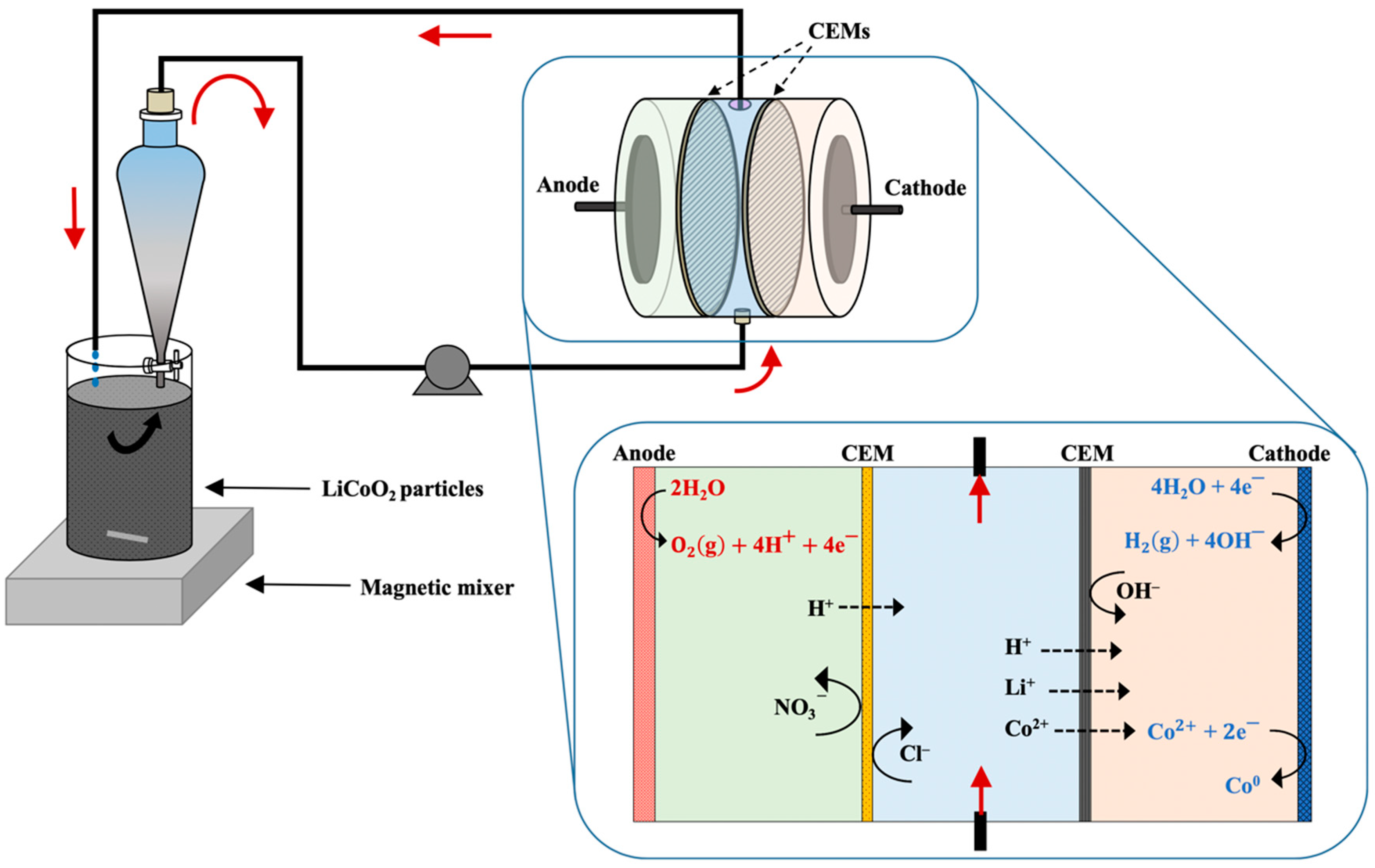
2.3. Hydrometallurgical–Electrodialytic Experiments
3. Results and Discussion
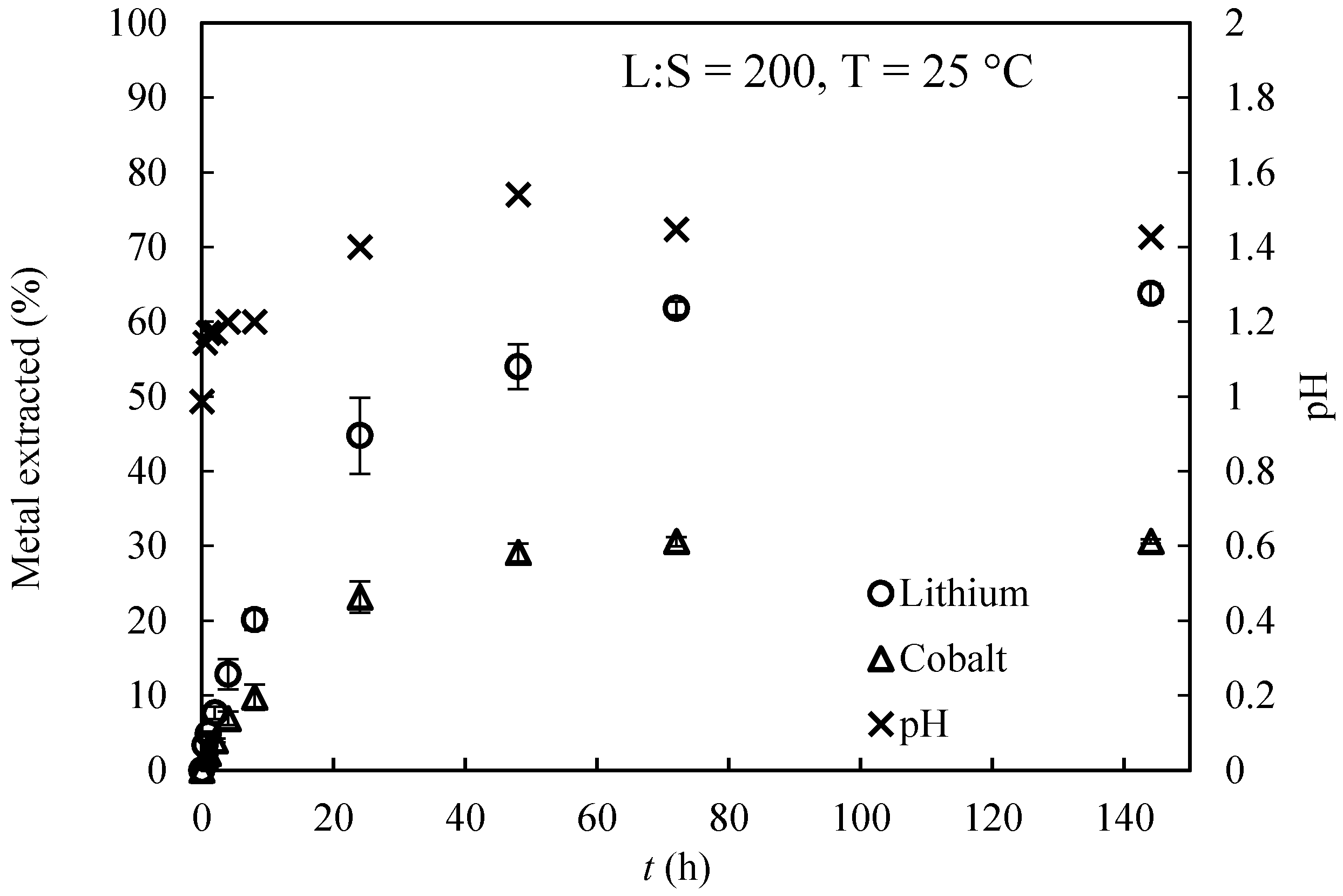
3.1. Extractions Eexperiments and Chemical Equilibrium Calculaitons
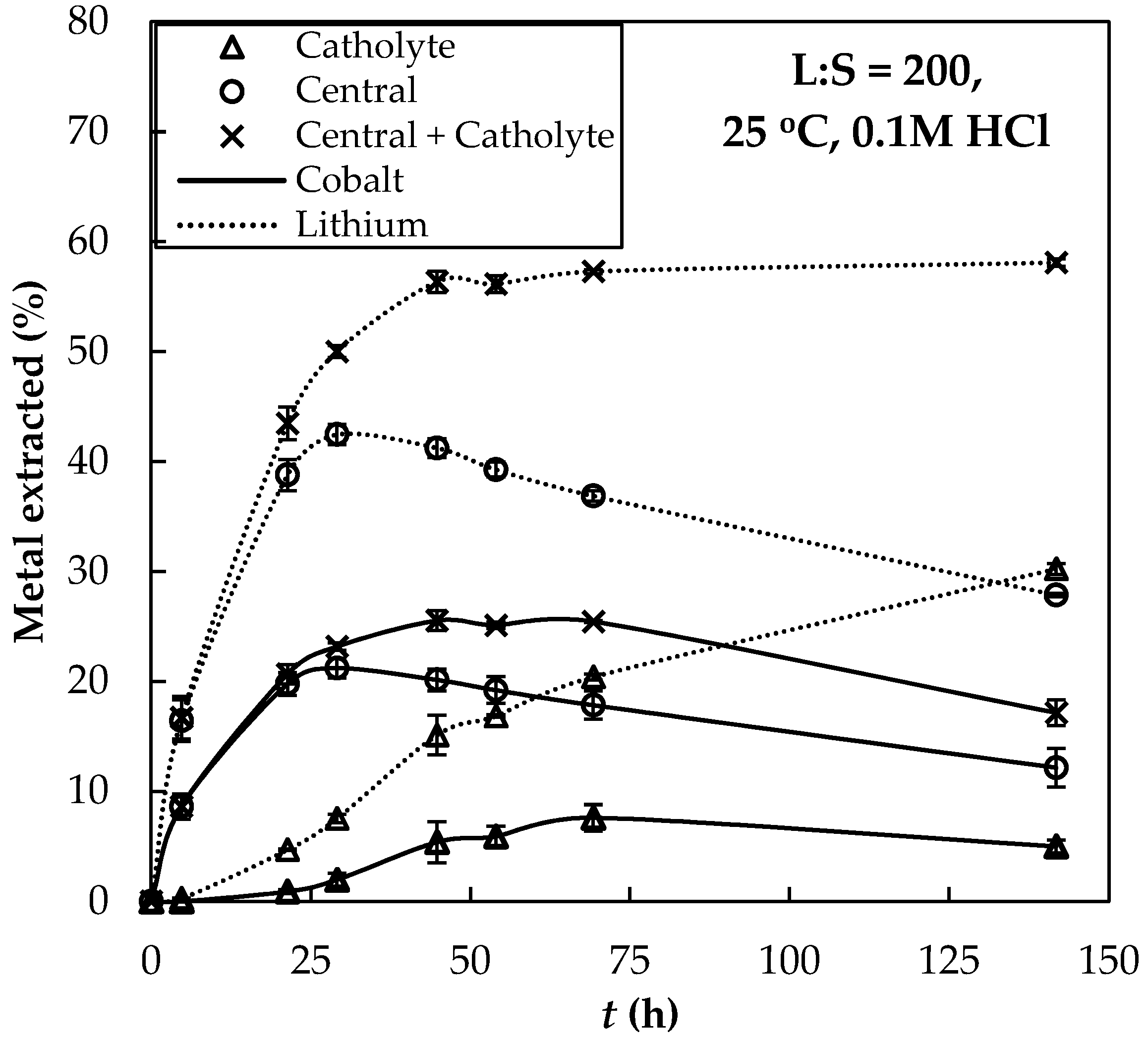
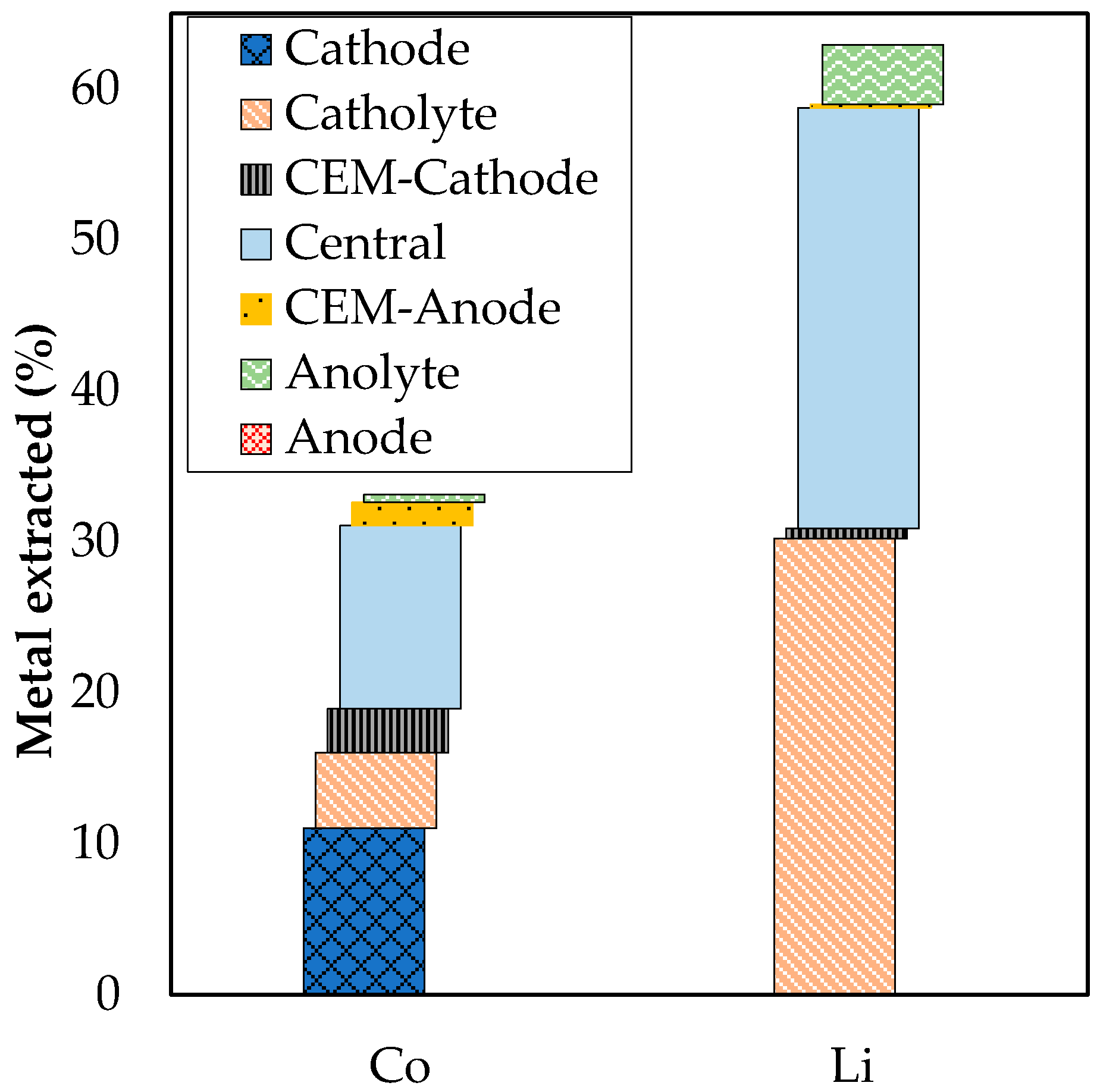
3.2. Extraction with ED
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zubi, G.; Dufo-López, R.; Carvalho, M.; Pasaoglu, G. The lithium-ion battery: State of the art and future perspectives. Renew. Sustain. Energy Rev. 2018, 89, 292–308. [Google Scholar] [CrossRef]
- Heelan, J.; Gratz, E.; Zheng, Z.; Wang, Y.; Chen, M.; Apelian, D. Current and prospective Li-Ion battery recycling and recovery processes. JOM 2016, 68, 2632–2638. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Z.; Lin, X.; Zhang, Y.; He, Y.; Cao, H.; Sun, Z. A mini-review on metal recycling from spent lithium ion batteries. Engineering 2018, 4, 361–370. [Google Scholar] [CrossRef]
- Melin, H.E. State of the Art in Reuse and Recycling of Lithium-Ion Batteries—A Research Review; The Swedish Energy Agency: London, UK, 2019. [Google Scholar]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Mathieux, F.; Ardente, F.; Bobba, S.; Nuss, P.; Blengini, G.A.; Dias, P.A.; Blagoeva, D.; De Matos, C.T.; Wittmer, D.; Pavel, C.; et al. Critical Raw Materials and the Circular Economy; Background report; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar] [CrossRef]
- Swain, B. Recovery and recycling of lithium: A review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Graedel, T.E.; Allwood, J.; Birat, J.P.; Buchert, M.; Hagelüken, C.; Reck, B.K.; Sibley, S.F.; Sonnemann, G. Recycling Rates of Metals: A Status Report; United Nations Environment Programme: Paris, France, 2011; ISBN 978-92-807-3161-3. [Google Scholar]
- Navarro-Suárez, A.; Casado, N.; Mecerreyes, D.; Rojo, T.; Castillo-Martínez, E.; Carretero-González, J. Hybrid biopolymer electrodes for lithium- and sodium-ion batteries in organic electrolytes. Sustain. Energy Fuels 2018, 2, 836–842. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2014, 7, 19–29. [Google Scholar] [CrossRef]
- Larsson, F.; Andersson, P.; Blomqvist, P.; Mellander, B.-E. Toxic fluoride gas emissions from lithium-ion battery fires. Sci. Rep. 2017, 7, 10018. [Google Scholar] [CrossRef]
- Chen, L.; Tang, X.; Zhang, Y.; Li, L.; Zeng, Z.; Zhang, Y. Process for the recovery of cobalt oxalate from spent lithium-ion batteries. Hydrometallurgy 2011, 108, 80–86. [Google Scholar] [CrossRef]
- Zeng, X.; Li, J.; Singh, N. Recycling of Spent Lithium-Ion Battery: A Critical Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 1129–1165. [Google Scholar] [CrossRef]
- Meshram, P.; Pandey, B.; Mankhand, T. Extraction of lithium from primary and secondary sources by pre-treatment, leaching and separation: A comprehensive review. Hydrometallurgy 2014, 150, 192–208. [Google Scholar] [CrossRef]
- Foster, M.; Isely, P.; Standridge, C.R.; Hasan, M. Feasibility assessment of remanufacturing, repurposing, and recycling of end of vehicle application lithium-ion batteries. J. Ind. Eng. Manag. 2014, 7, 698–715. [Google Scholar] [CrossRef]
- Choubey, P.K.; Chung, K.-S.; Kim, M.-S.; Lee, J.-C.L.; Srivastava, R.R. Advance review on the exploitation of the prominent energy-storage element Lithium. Part II: From sea water and spent lithium ion batteries (LIBs). Miner. Eng. 2017, 110, 104–121. [Google Scholar] [CrossRef]
- Villen-Guzman, M.; Arhoun, B.; Vereda-Alonso, C.; Gomez-Lahoz, C.; Rodriguez-Maroto, J.M.; Paz-Garcia, J.M. Electrodialytic processes in solid matrices. New insights into battery recycling. A review. J. Chem. Technol. Biotechnol. 2019, 94, 1727–1738. [Google Scholar] [CrossRef]
- Velizarova, E.; Ribeiro, A.B.; Ottosen, L.M. A comparative study on Cu, Cr and as removal from CCA-treated wood waste by dialytic and electrodialytic processes. J. Hazard. Mater. 2002, 94, 147–160. [Google Scholar] [CrossRef]
- Pedersen, A.J.; Ottosen, L.M.; Villumsen, A.; Damø, A.J. Electrodialytic removal of heavy metals from different fly ashes. J. Hazard. Mater. 2003, 100, 65–78. [Google Scholar] [CrossRef]
- Nystroem, G.M.; Ottosen, L.M.; Villumsen, A. Acidification of harbor sediment and removal of heavy metals induced by water splitting in electrodialytic remediation. Sep. Sci. Technol. 2005, 40, 2245–2264. [Google Scholar] [CrossRef]
- Ottosen, L.M.; Lepkova, K.; Kubal, M. Comparison of electrodialytic removal of Cu from spiked kaolinite, spiked soil and industrially polluted soil. J. Hazard. Mater. 2006, 137, 113–120. [Google Scholar] [CrossRef]
- Guedes, P.; Couto, N.; Ottosen, L.M.; Ribeiro, A.B. Phosphorus recovery from sewage sludge ash through an electrodialytic process. Waste Manag. 2014, 34, 886–892. [Google Scholar] [CrossRef]
- Huang, B.; Pan, Z.; Su, X.; An, L. Recycling of lithium-ion batteries: Recent advances and perspectives. J. Power Sources 2018, 399, 274–286. [Google Scholar] [CrossRef]
- Lee, C.K.; Rhee, K.-I. Preparation of LiCoO2 from spent lithium-ion batteries. J. Power Sources 2002, 109, 17–21. [Google Scholar] [CrossRef]
- Takacova, Z.; Havlik, T.; Kukurugya, F.; Orac, D. Cobalt and lithium recovery from active mass of spent Li-ion batteries: Theoretical and experimental approach. Hydrometallurgy 2016, 163, 9–17. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. In U.S. Geological Survey Techniques and Methods, Book 6, Chapter A43; U.S. Geological Survey: Reston, VA, USA, 2014. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Oxidation and Reduction; Equilibria and Microbial Mediation. In Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2012; pp. 425–515. [Google Scholar]
- Nakamura, T.; Kajiyama, A. Synthesis of LiCoO2 particles with uniform size distribution using hydrothermally precipitated Co3O4 fine particles. Solid State Ionics 1999, 123, 95–101. [Google Scholar] [CrossRef]
- Shuva, M.A.H.; Kurny, A.S.W. Dissolution Kinetics of Cathode of Spent Lithium Ion Battery in Hydrochloric Acid Solutions. J. Inst. Eng. (India) Ser. D 2013, 94, 13–16. [Google Scholar] [CrossRef]
- Li, L.; Bian, Y.; Zhang, X.; Guan, Y.; Fan, E.; Wu, F.; Chen, R. Process for recycling mixed-cathode materials from spent lithium-ion batteries and kinetics of leaching. Waste Manag. 2018, 71, 362–371. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation | |||
|---|---|---|---|
| Heterogeneous reactions | |||
| 52.307 | (1) | ||
| 35.293 | (2) | ||
| 5.265 | (3) | ||
| 46.378 | (4) | ||
| 30.724 | (5) | ||
| 12.2 | (6) | ||
| Redox half reactions | |||
| 1.920 | (7) | ||
| −0.28 | (8) | ||
| 1.358 | (9) | ||
| 1.482 | (10) | ||
| 1.229 | (11) | ||
| 0.695 | (12) | ||
| Homogeneous aqueous-phase reactions | |||
| 10.6 | (13) | ||
| 1.512 | (14) | ||
| 0.710 | (15) | ||
| −0.570 | (16) | ||
| −0.020 | (17) | ||
| 1.710 | (18) | ||
| 2.090 | (19) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerrillo-Gonzalez, M.M.; Villen-Guzman, M.; Vereda-Alonso, C.; Gomez-Lahoz, C.; Rodriguez-Maroto, J.M.; Paz-Garcia, J.M. Recovery of Li and Co from LiCoO2 via Hydrometallurgical–Electrodialytic Treatment. Appl. Sci. 2020, 10, 2367. https://doi.org/10.3390/app10072367
Cerrillo-Gonzalez MM, Villen-Guzman M, Vereda-Alonso C, Gomez-Lahoz C, Rodriguez-Maroto JM, Paz-Garcia JM. Recovery of Li and Co from LiCoO2 via Hydrometallurgical–Electrodialytic Treatment. Applied Sciences. 2020; 10(7):2367. https://doi.org/10.3390/app10072367
Chicago/Turabian StyleCerrillo-Gonzalez, M.M., M. Villen-Guzman, C. Vereda-Alonso, C. Gomez-Lahoz, J.M. Rodriguez-Maroto, and J.M. Paz-Garcia. 2020. "Recovery of Li and Co from LiCoO2 via Hydrometallurgical–Electrodialytic Treatment" Applied Sciences 10, no. 7: 2367. https://doi.org/10.3390/app10072367
APA StyleCerrillo-Gonzalez, M. M., Villen-Guzman, M., Vereda-Alonso, C., Gomez-Lahoz, C., Rodriguez-Maroto, J. M., & Paz-Garcia, J. M. (2020). Recovery of Li and Co from LiCoO2 via Hydrometallurgical–Electrodialytic Treatment. Applied Sciences, 10(7), 2367. https://doi.org/10.3390/app10072367