Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs

Abstract
1. Introduction
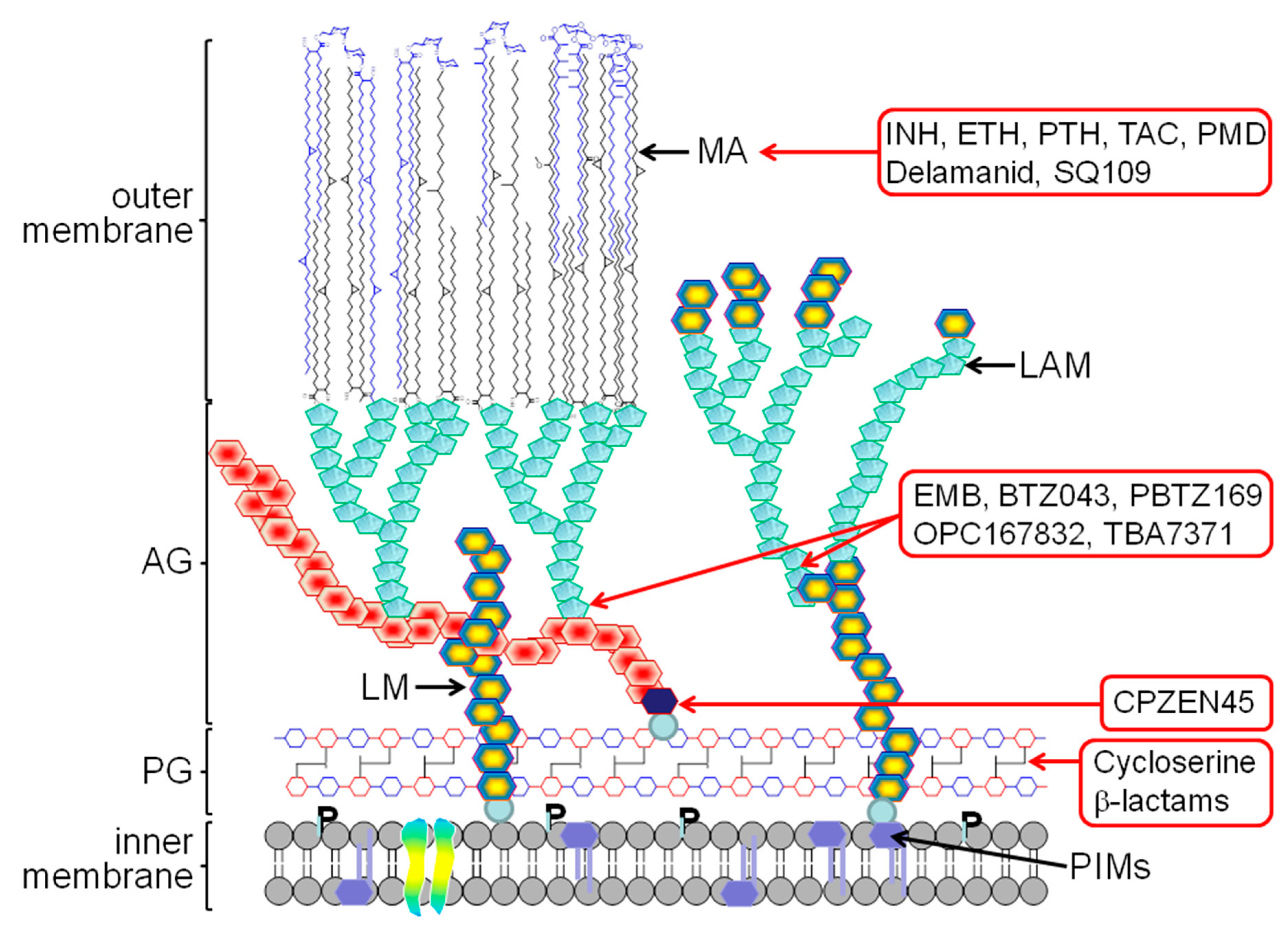
2. The Mycobacterial Cell Wall
3. Old TB Drugs
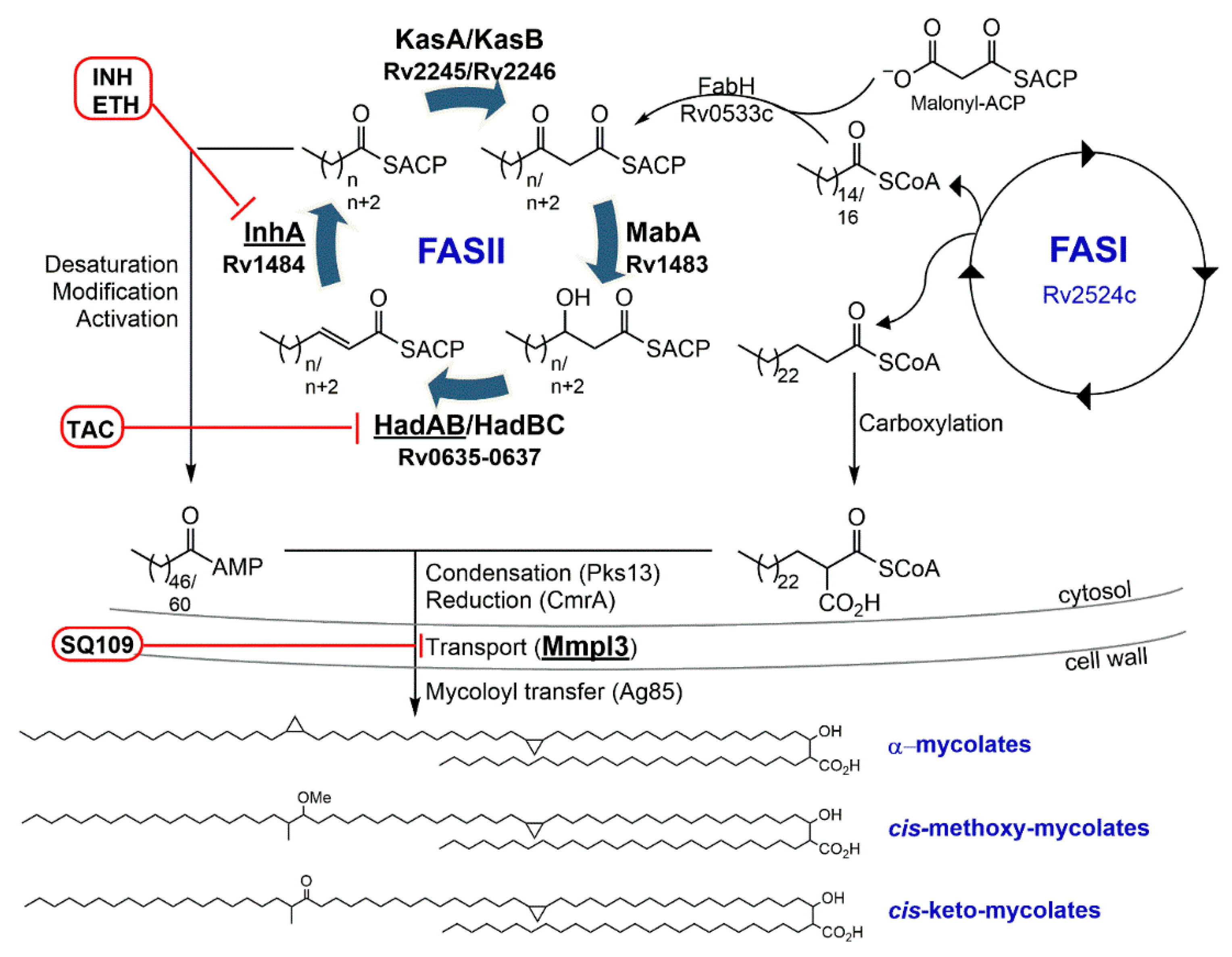
3.1. Isoniazid—Target: Mycolic Acids
3.2. Ethambutol—Target: Arabinogalactan/LAM
3.3. Ethionamide/Prothionamide—Target: Mycolic Acids
3.4. Cycloserine—Target: Peptidoglycan
3.5. Thiacetazone—Target: Mycolic Acids
4. New Generation of Cell Wall Inhibitors
4.1. Target: Mycolic Acids
4.1.1. SQ109
4.1.2. Pretomanid (PA-824)
4.1.3. Delamanid (OPC-67683)
4.2. Target: Arabinogalactan/LAM
4.2.1. CPZEN-45
4.2.2. BTZ043
4.2.3. PBTZ169
4.2.4. OPC167832
4.2.5. TBA-7371
4.3. Target: Peptidoglycan
4.3.1. β-Lactams and Clavulanic Acid
4.3.2. Sanfetrinem
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Daniel, T.M. The history of tuberculosis. Respir. Med. 2006, 100, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Keers, R.Y. Two forgotten pioneers. James Carson and George Bodington. Thorax 1980, 35, 483–489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bodington, G. Reviews of Books. An essay on the treatment and cure of pulmunory consumption, on principles natural, ratinal and succesful. Lancet 1840, 34, 575–577. [Google Scholar]
- George Bodington, M.D. Obituary. Lancet 1882, 119, 477. [Google Scholar]
- McCarthy, O.R. The key to the sanatoria. J. R. Soc. Med. 2001, 94, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Latour, A. Review of Books. On the preservative and curative treatment of pulmunory consumption. Lancet 1840, 34, 616–619. [Google Scholar]
- Cox, G.L. Sanatorium treatment contrasted with home treatment. After-histories of 4067 cases. Br. J. Tuberc. 1923, 17, 27–30. [Google Scholar]
- Schatz, A.; Bugie, E.; Waksman, S.A. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc. Exp. Biol. Med. 1944, 55, 66–69. [Google Scholar] [CrossRef]
- Schatz, A.; Waksman, S.A. Effect of streptomycin upon Mycobacterium tuberculosis and related organisms. Proc. Soc. Exp. Biol. Med. 1944, 57, 244–248. [Google Scholar] [CrossRef]
- Pfuetze, K.H.; Pyle, M.M.; Hinshaw, H.C.; Feldman, W.H. The first clinical trial of streptomycin in human tuberculosis. Am. Rev. Tuberc. 1955, 71, 752–754. [Google Scholar] [CrossRef]
- Waksman, S.A. Tenth anniversary of the discovery of streptomycin, the first chemotherapeutic agent found to be effective against tuberculosis in humans. Am. Rev. Tuberc. 1954, 70, 1–8. [Google Scholar] [CrossRef]
- Hinshaw, H.C.; Pyle, M.M.; Feldman, W.H. Streptomycin in tuberculosis. Am. J. Med. 1947, 2, 429–435. [Google Scholar] [CrossRef]
- Crofton, J.; Mitchison, D.A. Streptomycin resistance in pulmonary tuberculosis. Br. Med. J. 1948, 2, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J. Para-Aminosalicylic acid in the treatment of tuberculosis. Lancet 1946, 247, 15. [Google Scholar] [CrossRef]
- Medical Research Council Investigation. Treatment of pulmonary tuberculosis with streptomycin and para-amino-salicylic acid. Br. Med. J. 1950, 2, 4688. [Google Scholar]
- Crofton, J. Chemotherapy of pulmonary tuberculosis. Br. Med. J. 1959, 1, 1610–1614. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1 (accessed on 15 February 2020).
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: A multifaceted antibiotic target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef]
- Konyarikova, Z.; Savkova, K.; Kozmon, S.; Mikusova, K. Biosynthesis of Galactan in Mycobacterium tuberculosis as a Viable TB Drug Target? Antibiotics 2020, 9, 20. [Google Scholar] [CrossRef]
- Maitra, A.; Munshi, T.; Healy, J.; Martin, L.T.; Vollmer, W.; Keep, N.H.; Bhakta, S. Cell wall peptidoglycan in Mycobacterium tuberculosis: An Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 2019, 43, 548–575. [Google Scholar] [CrossRef] [PubMed]
- Lederer, E.; Adam, A.; Ciorbaru, R.; Petit, J.F.; Wietzerbin, J. Cell walls of Mycobacteria and related organisms; chemistry and immunostimulant properties. Mol. Cell. Biochem. 1975, 7, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Crick, D.C.; Brennan, P.J. Comparison of the UDP-N-acetylmuramate:L-alanine ligase enzymes from Mycobacterium tuberculosis and Mycobacterium leprae. J. Bacteriol. 2000, 182, 6827–6830. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Scherman, H.; Brennan, P.J.; Crick, D.C. N-Glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J. Bacteriol. 2005, 187, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.B.; Mahapatra, S.; Crick, D.C.; Pavelka, M.S., Jr. Identification of the namH gene, encoding the hydroxylase responsible for the N-glycolylation of the mycobacterial peptidoglycan. J. Biol. Chem. 2005, 280, 326–333. [Google Scholar] [CrossRef]
- McNeil, M.; Wallner, S.J.; Hunter, S.W.; Brennan, P.J. Demonstration that the galactosyl and arabinosyl residues in the cell-wall arabinogalactan of Mycobacterium leprae and Mycobacterium tuberculosis are furanoid. Carbohydr. Res. 1987, 166, 299–308. [Google Scholar] [CrossRef]
- McNeil, M.; Daffe, M.; Brennan, P.J. Evidence for the nature of the link between the arabinogalactan and peptidoglycan of mycobacterial cell walls. J. Biol. Chem. 1990, 265, 18200–18206. [Google Scholar]
- Marrakchi, H.; Laneelle, M.A.; Daffe, M. Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef]
- Brindley, D.N.; Matsumura, S.; Bloch, K. Mycobacterium phlei Fatty Acid Synthetase—A Bacterial Multienzyme Complex. Nature 1969, 224, 666–669. [Google Scholar] [CrossRef]
- Cantaloube, S.; Veyron-Churlet, R.; Haddache, N.; Daffe, M.; Zerbib, D. The Mycobacterium tuberculosis FAS-II dehydratases and methyltransferases define the specificity of the mycolic acid elongation complexes. PLoS ONE 2011, 6, e29564. [Google Scholar] [CrossRef]
- Fukuda, T.; Matsumura, T.; Ato, M.; Hamasaki, M.; Nishiuchi, Y.; Murakami, Y.; Maeda, Y.; Yoshimori, T.; Matsumoto, S.; Kobayashi, K.; et al. Critical roles for lipomannan and lipoarabinomannan in cell wall integrity of mycobacteria and pathogenesis of tuberculosis. MBio 2013, 4, e00472-12. [Google Scholar] [CrossRef] [PubMed]
- Nigou, J.; Gilleron, M.; Rojas, M.; Garcia, L.F.; Thurnher, M.; Puzo, G. Mycobacterial lipoarabinomannans: Modulators of dendritic cell function and the apoptotic response. Microbes Infect. 2002, 4, 945–953. [Google Scholar] [CrossRef]
- Patterson, J.H.; Waller, R.F.; Jeevarajah, D.; Billman-Jacobe, H.; McConville, M.J. Mannose metabolism is required for mycobacterial growth. Biochem. J. 2003, 372, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Cox, J.S. Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr. Top. Microbiol. Immunol. 2013, 374, 211–241. [Google Scholar] [CrossRef] [PubMed]
- McDermott, W. The story of INH. J. Infect. Dis. 1969, 119, 678–683. [Google Scholar] [CrossRef]
- Hinshaw, H.C.; Mc, D.W. Thiosemicarbazone therapy of tuberculosis in humans. Am. Rev. Tuberc. 1950, 61, 145–157. [Google Scholar]
- Donovick, R.; Pansy, F.; Stryker, G.; Bernstein, J. The chemotherapy of experimental tuberculosis. I. The in vitro activity of thiosemicarbazides, thiosemicarbazones, and related compounds. J. Bacteriol. 1950, 59, 667–674. [Google Scholar] [CrossRef]
- Fox, H.H. The chemical approach to the control of tuberculosis. Science 1952, 116, 129–134. [Google Scholar] [CrossRef]
- Hamre, D.; Bernstein, J.; Donovick, R. The chemotherapy of experimental tuberculosis. II. Thiosemicarbazones and analogues in experimental tuberculosis in the mouse. J. Bacteriol. 1950, 59, 675–680. [Google Scholar] [CrossRef]
- Bernstein, J.W.; Lott, A.; Steinberg, B.A.; Yale, H.L. Chemotherapy of experimental tuberculosis. Am. Rev. Tuberc. 1952, 65, 357–374. [Google Scholar]
- Domagk, G.; Offe, H.A.; Siefken, W. [Therapy of experimental tuberculosis with neoteben]. Med. Colon. 1952, 20, 517–528. [Google Scholar] [PubMed]
- Jain, P.; Weinrick, B.C.; Kalivoda, E.J.; Yang, H.; Munsamy, V.; Vilcheze, C.; Weisbrod, T.R.; Larsen, M.H.; O’Donnell, M.R.; Pym, A.; et al. Dual-Reporter Mycobacteriophages (Phi2DRMs) Reveal Preexisting Mycobacterium tuberculosis Persistent Cells in Human Sputum. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, P.C.; Williams, E.P.; Bishai, W.R. Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2008, 61, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Yamaguchi, I.; Yamamoto, I.; Fukuda, T.; Yokota, S.; Maekura, R.; Ito, M.; Yamamoto, Y.; Ogura, T.; Maeda, K.; et al. Slow N-acetyltransferase 2 genotype affects the incidence of isoniazid and rifampicin-induced hepatotoxicity. Int. J. Tuberc. Lung Dis. 2000, 4, 256–261. [Google Scholar] [PubMed]
- Wang, P.; Pradhan, K.; Zhong, X.B.; Ma, X. Isoniazid metabolism and hepatotoxicity. Acta Pharm. Sin. B 2016, 6, 384–392. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Zimmerman, H.J.; Ishak, K.G.; Thorgeirsson, U.P.; Timbrell, J.A.; Snodgrass, W.R.; Nelson, S.D. Isoniazid liver injury: Clinical spectrum, pathology, and probable pathogenesis. Ann. Intern. Med. 1976, 84, 181–192. [Google Scholar] [CrossRef]
- Nelson, S.D.; Mitchell, J.R.; Timbrell, J.A.; Snodgrass, W.R.; Corcoran, G.B., 3rd. Isoniazid and iproniazid: Activation of metabolites to toxic intermediates in man and rat. Science 1976, 193, 901–903. [Google Scholar] [CrossRef]
- Lauterburg, B.H.; Smith, C.V.; Todd, E.L.; Mitchell, J.R. Pharmacokinetics of the toxic hydrazino metabolites formed from isoniazid in humans. J. Pharmacol. Exp. Ther. 1985, 235, 566–570. [Google Scholar]
- Gajdacs, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef]
- Laborde, J.; Deraeve, C.; Bernardes-Genisson, V. Update of Antitubercular Prodrugs from a Molecular Perspective: Mechanisms of Action, Bioactivation Pathways, and Associated Resistance. ChemMedChem 2017, 12, 1657–1676. [Google Scholar] [CrossRef]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, K.; Schultz, P.G. Mechanistic Studies of the Oxidation of Isoniazid by the Catalase Peroxidase from Mycobacterium tuberculosis. J. Am. Chem. Soc. 1994, 116, 7425–7426. [Google Scholar] [CrossRef]
- Lei, B.; Wei, C.J.; Tu, S.C. Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of inhA inhibitor. J. Biol. Chem. 2000, 275, 2520–2526. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R., Jr.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef]
- Wilming, M.; Johnsson, K. Spontaneous Formation of the Bioactive Form of the Tuberculosis Drug Isoniazid. Angew. Chem. Int. Ed. Engl. 1999, 38, 2588–2590. [Google Scholar] [CrossRef]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R., Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef]
- Nguyen, M.; Quemard, A.; Broussy, S.; Bernadou, J.; Meunier, B. Mn(III) pyrophosphate as an efficient tool for studying the mode of action of isoniazid on the InhA protein of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2137–2144. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef]
- Vilcheze, C.; Wang, F.; Arai, M.; Hazbon, M.H.; Colangeli, R.; Kremer, L.; Weisbrod, T.R.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R., Jr. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 2006, 12, 1027–1029. [Google Scholar] [CrossRef]
- Dessen, A.; Quemard, A.; Blanchard, J.S.; Jacobs, W.R., Jr.; Sacchettini, J.C. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 1995, 267, 1638–1641. [Google Scholar] [CrossRef]
- Quemard, A.; Sacchettini, J.C.; Dessen, A.; Vilcheze, C.; Bittman, R.; Jacobs, W.R., Jr.; Blanchard, J.S. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995, 34, 8235–8241. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Laneelle, G.; Quemard, A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 2000, 146 Pt 2, 289–296. [Google Scholar] [CrossRef]
- Takayama, K.; Wang, L.; David, H.L. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1972, 2, 29–35. [Google Scholar] [CrossRef]
- Vilcheze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R., Jr. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef]
- Winder, F.G.; Collins, P.B. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 1970, 63, 41–48. [Google Scholar] [CrossRef]
- Vilcheze, C.; Jacobs, W.R., Jr. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Middlebrook, G.; Cohn, M.L. Some observations on the pathogenicity of isoniazid-resistant variants of tubercle bacilli. Science 1953, 118, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Middlebrook, G. Isoniazid resistance and catalase activity of tubercle bacilli. Am. Rev. Tuberc. 1954, 69, 471–472. [Google Scholar] [PubMed]
- Zhang, Y.; Garbe, T.; Young, D. Transformation with katG restores isoniazid-sensitivity in Mycobacterium tuberculosis isolates resistant to a range of drug concentrations. Mol. Microbiol. 1993, 8, 521–524. [Google Scholar] [CrossRef]
- Vilcheze, C.; Jacobs, W.R., Jr. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: Genes, Mutations, and Causalities. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Ando, H.; Kondo, Y.; Suetake, T.; Toyota, E.; Kato, S.; Mori, T.; Kirikae, T. Identification of katG mutations associated with high-level isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, J.; Miyoshi-Akiyama, T.; Augustynowicz-Kopec, E.; Zwolska, Z.; Kirikae, F.; Toyota, E.; Kobayashi, I.; Morita, K.; Kudo, K.; Kato, S.; et al. Detection of multidrug resistance in Mycobacterium tuberculosis. J. Clin. Microbiol. 2007, 45, 179–192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, C.J.; Lei, B.; Musser, J.M.; Tu, S.C. Isoniazid activation defects in recombinant Mycobacterium tuberculosis catalase-peroxidase (KatG) mutants evident in InhA inhibitor production. Antimicrob. Agents Chemother. 2003, 47, 670–675. [Google Scholar] [CrossRef]
- Brossier, F.; Veziris, N.; Truffot-Pernot, C.; Jarlier, V.; Sougakoff, W. Performance of the genotype MTBDR line probe assay for detection of resistance to rifampin and isoniazid in strains of Mycobacterium tuberculosis with low- and high-level resistance. J. Clin. Microbiol. 2006, 44, 3659–3664. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Kitao, T.; Miyoshi-Akiyama, T.; Kato, S.; Mori, T.; Kirikae, T. Downregulation of katG expression is associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 2011, 79, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Streicher, E.M.; Hoek, K.G.; Tait, M.; Trollip, A.; Bosman, M.E.; Coetzee, G.J.; Chabula-Nxiweni, E.M.; Hoosain, E.; Gey van Pittius, N.C.; et al. inhA promoter mutations: A gateway to extensively drug-resistant tuberculosis in South Africa? Int. J. Tuberc. Lung Dis. 2011, 15, 344–351. [Google Scholar] [PubMed]
- Shaw, D.J.; Robb, K.; Vetter, B.V.; Tong, M.; Molle, V.; Hunt, N.T.; Hoskisson, P.A. Disruption of key NADH-binding pocket residues of the Mycobacterium tuberculosis InhA affects DD-CoA binding ability. Sci. Rep. 2017, 7, 4714. [Google Scholar] [CrossRef]
- Machado, D.; Perdigao, J.; Ramos, J.; Couto, I.; Portugal, I.; Ritter, C.; Boettger, E.C.; Viveiros, M. High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J Antimicrob. Chemother. 2013, 68, 1728–1732. [Google Scholar] [CrossRef]
- Morlock, G.P.; Metchock, B.; Sikes, D.; Crawford, J.T.; Cooksey, R.C. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2003, 47, 3799–3805. [Google Scholar] [CrossRef]
- Nimmo, C.; Doyle, R.; Burgess, C.; Williams, R.; Gorton, R.; McHugh, T.D.; Brown, M.; Morris-Jones, S.; Booth, H.; Breuer, J. Rapid identification of a Mycobacterium tuberculosis full genetic drug resistance profile through whole genome sequencing directly from sputum. Int. J. Infect. Dis. 2017, 62, 44–46. [Google Scholar] [CrossRef]
- Encinas, L.; O’Keefe, H.; Neu, M.; Remuinan, M.J.; Patel, A.M.; Guardia, A.; Davie, C.P.; Perez-Macias, N.; Yang, H.; Convery, M.A.; et al. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Sala, C.; Neres, J.; Pojer, F.; Magnet, S.; Mukherjee, R.; Uplekar, S.; Boy-Rottger, S.; Altmann, K.H.; Cole, S.T. Towards a new tuberculosis drug: Pyridomycin—Nature’s isoniazid. EMBO Mol. Med. 2012, 4, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; You, Q.D.; Chen, Y.D. Recent progress in the identification and development of InhA direct inhibitors of Mycobacterium tuberculosis. Mini Rev. Med. Chem. 2010, 10, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; SP, S.R.; Kondreddi, R.R.; Noble, C.G.; Camacho, L.R.; Tan, B.H.; Ng, S.H.; Ng, P.S.; Ma, N.L.; Lakshminarayana, S.B.; et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015, 7, 269ra263. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Tonge, P.J. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr. Top. Med. Chem. 2012, 12, 672–693. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef]
- Sink, R.; Sosic, I.; Zivec, M.; Fernandez-Menendez, R.; Turk, S.; Pajk, S.; Alvarez-Gomez, D.; Lopez-Roman, E.M.; Gonzales-Cortez, C.; Rullas-Triconado, J.; et al. Design, synthesis, and evaluation of new thiadiazole-based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J. Med. Chem. 2015, 58, 613–624. [Google Scholar] [CrossRef]
- Vilcheze, C.; Baughn, A.D.; Tufariello, J.; Leung, L.W.; Kuo, M.; Basler, C.F.; Alland, D.; Sacchettini, J.C.; Freundlich, J.S.; Jacobs, W.R., Jr. Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrob. Agents Chemother. 2011, 55, 3889–3898. [Google Scholar] [CrossRef]
- Martinez-Hoyos, M.; Perez-Herran, E.; Gulten, G.; Encinas, L.; Alvarez-Gomez, D.; Alvarez, E.; Ferrer-Bazaga, S.; Garcia-Perez, A.; Ortega, F.; Angulo-Barturen, I.; et al. Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 2016, 8, 291–301. [Google Scholar] [CrossRef]
- Robertson, G.T.; Ektnitphong, V.A.; Scherman, M.S.; McNeil, M.B.; Dennison, D.; Korkegian, A.; Smith, A.J.; Halladay, J.; Carter, D.S.; Xia, Y.; et al. Efficacy and Improved Resistance Potential of a Cofactor-Independent InhA Inhibitor of Mycobacterium tuberculosis in the C3HeB/FeJ Mouse Model. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Xia, Y.; Zhou, Y.; Carter, D.S.; McNeil, M.B.; Choi, W.; Halladay, J.; Berry, P.W.; Mao, W.; Hernandez, V.; O’Malley, T.; et al. Discovery of a cofactor-independent inhibitor of Mycobacterium tuberculosis InhA. Life Sci. Alliance 2018, 1, e201800025. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.G.; Wilkinson, R.G. Antituberculous Agents. Ii. N,N′-Diisopropylethylenediamine and Analogs. J. Med. Chem. 1962, 91, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.P.; Baughn, C.O.; Wilkinson, R.G.; Shepherd, R.G. A new synthetic compound with antituberculous activity in mice: Ethambutol (dextro-2,2′-(ethylenediimino)-di-l-butanol). Am. Rev. Respir. Dis. 1961, 83, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.G.; Shepherd, R.G.; Thomas, J.P.; Baughn, C.O. Stereospecificity in a new type of synthetic antituberculous agent. J. Am. Chem. Soc. 1961, 83, 2212. [Google Scholar] [CrossRef]
- Kuck, N.A.; Peets, E.A.; Forbes, M. Mode of action of ethambutol on Mycobacterium tuberculosis, strain H37R V. Am. Rev. Respir. Dis. 1963, 87, 905–906. [Google Scholar] [CrossRef]
- Koul, P.A. Ocular toxicity with ethambutol therapy: Timely recaution. Lung India 2015, 32, 1–3. [Google Scholar] [CrossRef]
- Forbes, M.; Kuck, N.A.; Peets, E.A. Effect of Ethambutol on Nucleic Acid Metabolism in Mycobacterium Smegmatis and Its Reversal by Polyamines and Divalent Cations. J. Bacteriol. 1965, 89, 1299–1305. [Google Scholar] [CrossRef]
- Takayama, K.; Armstrong, E.L.; Kunugi, K.A.; Kilburn, J.O. Inhibition by ethambutol of mycolic acid transfer into the cell wall of Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1979, 16, 240–242. [Google Scholar] [CrossRef]
- Kilburn, J.O.; Takayama, K. Effects of ethambutol on accumulation and secretion of trehalose mycolates and free mycolic acid in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1981, 20, 401–404. [Google Scholar] [CrossRef]
- Takayama, K.; Kilburn, J.O. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. [Google Scholar] [CrossRef]
- Wolucka, B.A.; McNeil, M.R.; de Hoffmann, E.; Chojnacki, T.; Brennan, P.J. Recognition of the lipid intermediate for arabinogalactan/arabinomannan biosynthesis and its relation to the mode of action of ethambutol on mycobacteria. J. Biol. Chem. 1994, 269, 23328–23335. [Google Scholar] [PubMed]
- Mikusova, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Mikusova, K.; Robuck, K.G.; Scherman, M.; Brennan, P.J.; McNeil, M.R. Recognition of multiple effects of ethambutol on metabolism of mycobacterial cell envelope. Antimicrob. Agents Chemother. 1995, 39, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Mikusova, K.; Slayden, R.A.; Besra, G.S.; Brennan, P.J. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 1995, 39, 2484–2489. [Google Scholar] [CrossRef]
- Pawar, A.; Jha, P.; Konwar, C.; Chaudhry, U.; Chopra, M.; Saluja, D. Ethambutol targets the glutamate racemase of Mycobacterium tuberculosis-an enzyme involved in peptidoglycan biosynthesis. Appl. Microbiol. Biotechnol. 2019, 103, 843–851. [Google Scholar] [CrossRef]
- Sreevatsan, S.; Stockbauer, K.E.; Pan, X.; Kreiswirth, B.N.; Moghazeh, S.L.; Jacobs, W.R., Jr.; Telenti, A.; Musser, J.M. Ethambutol resistance in Mycobacterium tuberculosis: Critical role of embB mutations. Antimicrob. Agents Chemother. 1997, 41, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Philipp, W.J.; Sreevatsan, S.; Bernasconi, C.; Stockbauer, K.E.; Wieles, B.; Musser, J.M.; Jacobs, W.R., Jr. The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 1997, 3, 567–570. [Google Scholar] [CrossRef]
- Belanger, A.E.; Besra, G.S.; Ford, M.E.; Mikusova, K.; Belisle, J.T.; Brennan, P.J.; Inamine, J.M. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. USA 1996, 93, 11919–11924. [Google Scholar] [CrossRef]
- Safi, H.; Sayers, B.; Hazbon, M.H.; Alland, D. Transfer of embB codon 306 mutations into clinical Mycobacterium tuberculosis strains alters susceptibility to ethambutol, isoniazid, and rifampin. Antimicrob. Agents Chemother. 2008, 52, 2027–2034. [Google Scholar] [CrossRef]
- Starks, A.M.; Gumusboga, A.; Plikaytis, B.B.; Shinnick, T.M.; Posey, J.E. Mutations at embB codon 306 are an important molecular indicator of ethambutol resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 1061–1066. [Google Scholar] [CrossRef]
- Hazbon, M.H.; Bobadilla del Valle, M.; Guerrero, M.I.; Varma-Basil, M.; Filliol, I.; Cavatore, M.; Colangeli, R.; Safi, H.; Billman-Jacobe, H.; Lavender, C.; et al. Role of embB codon 306 mutations in Mycobacterium tuberculosis revisited: A novel association with broad drug resistance and IS6110 clustering rather than ethambutol resistance. Antimicrob. Agents Chemother. 2005, 49, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Othman, S.N.; Ho, Y.M.; Wong, S.Y. Novel mutations within the embB gene in ethambutol-susceptible clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2004, 48, 4447–4449. [Google Scholar] [CrossRef] [PubMed]
- Mokrousov, I.; Otten, T.; Vyshnevskiy, B.; Narvskaya, O. Detection of embB306 mutations in ethambutol-susceptible clinical isolates of Mycobacterium tuberculosis from Northwestern Russia: Implications for genotypic resistance testing. J. Clin. Microbiol. 2002, 40, 3810–3813. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Shen, G.M.; Wu, J.; Gui, X.H.; Li, X.; Mei, J.; DeRiemer, K.; Gao, Q. Association between embB codon 306 mutations and drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 2618–2620. [Google Scholar] [CrossRef]
- Safi, H.; Lingaraju, S.; Amin, A.; Kim, S.; Jones, M.; Holmes, M.; McNeil, M.; Peterson, S.N.; Chatterjee, D.; Fleischmann, R.; et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 2013, 45, 1190–1197. [Google Scholar] [CrossRef]
- Tulyaprawat, O.; Chaiprasert, A.; Chongtrakool, P.; Suwannakarn, K.; Ngamskulrungroj, P. Association of ubiA mutations and high-level of ethambutol resistance among Mycobacterium tuberculosis Thai clinical isolates. Tuberculosis 2019, 114, 42–46. [Google Scholar] [CrossRef]
- Gardner, T.S.; Wenis, E.; Lee, J.H. The synthesis of compounds for the chemotherapy of tuberculosis. IV. The amide function. J. Org. Chem. 1954, 19, 753–757. [Google Scholar] [CrossRef]
- Grumbach, F.; Rist, N.; Libermann, D.; Moyeux, M.; Cals, S.; Clavel, S. Experimental antituberculous activity of certain isonicotinic thioamides substituted on the nucleus. C. R. Hebd. Seances Acad. Sci. 1956, 242, 2187–2189. [Google Scholar]
- Scardigli, A.; Caminero, J.A.; Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Migliori, G.B. Efficacy and tolerability of ethionamide versus prothionamide: A systematic review. Eur. Respir. J. 2016, 48, 946–952. [Google Scholar] [CrossRef]
- Winder, F.G.; Collins, P.B.; Whelan, D. Effects of ethionamide and isoxyl on mycolic acid synthesis in Mycobacterium tuberculosis BCG. J. Gen. Microbiol. 1971, 66, 379–380. [Google Scholar] [CrossRef][Green Version]
- Hok, T.T. A comparative study of the susceptibility to ethionamide, thiosemicarbazone, and isoniazid of tubercle bacilli from patients never treated with ethionamide or thiosemicarbazone. Am. Rev. Respir. Dis. 1964, 90, 468–469. [Google Scholar] [PubMed]
- Lefford, M.J. The ethionamide sensitivity of British pre-treatment strains of Mycobacterium tuberculosis. Tubercle 1966, 47, 198–206. [Google Scholar] [CrossRef]
- Stewart, S.M.; Hall, E.; Riddell, R.W.; Somner, A.R. Bacteriological aspects of the use of ethionamide, pyrazinamide and cycloserine in the treatment of chronic pulmonary tuberculosis. Tubercle 1962, 43, 417–431. [Google Scholar] [CrossRef]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.G.; Barry, C.E., 3rd. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9682. [Google Scholar] [CrossRef]
- Vannelli, T.A.; Dykman, A.; Ortiz de Montellano, P.R. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J. Biol. Chem. 2002, 277, 12824–12829. [Google Scholar] [CrossRef]
- Wang, F.; Langley, R.; Gulten, G.; Dover, L.G.; Besra, G.S.; Jacobs, W.R., Jr.; Sacchettini, J.C. Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 2007, 204, 73–78. [Google Scholar] [CrossRef]
- Ang, M.L.T.; Zainul Rahim, S.Z.; de Sessions, P.F.; Lin, W.; Koh, V.; Pethe, K.; Hibberd, M.L.; Alonso, S. EthA/R-Independent Killing of Mycobacterium tuberculosis by Ethionamide. Front. Microbiol. 2017, 8, 710. [Google Scholar] [CrossRef]
- Grant, S.S.; Wellington, S.; Kawate, T.; Desjardins, C.A.; Silvis, M.R.; Wivagg, C.; Thompson, M.; Gordon, K.; Kazyanskaya, E.; Nietupski, R.; et al. Baeyer-Villiger Monooxygenases EthA and MymA Are Required for Activation of Replicating and Non-replicating Mycobacterium tuberculosis Inhibitors. Cell Chem. Biol. 2016, 23, 666–677. [Google Scholar] [CrossRef]
- Hicks, N.D.; Carey, A.F.; Yang, J.; Zhao, Y.; Fortune, S.M. Bacterial Genome-Wide Association Identifies Novel Factors That Contribute to Ethionamide and Prothionamide Susceptibility in Mycobacterium tuberculosis. MBio 2019, 10. [Google Scholar] [CrossRef]
- Brossier, F.; Veziris, N.; Truffot-Pernot, C.; Jarlier, V.; Sougakoff, W. Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 355–360. [Google Scholar] [CrossRef]
- Zhang, H.N.; Xu, Z.W.; Jiang, H.W.; Wu, F.L.; He, X.; Liu, Y.; Guo, S.J.; Li, Y.; Bi, L.J.; Deng, J.Y.; et al. Cyclic di-GMP regulates Mycobacterium tuberculosis resistance to ethionamide. Sci. Rep. 2017, 7, 5860. [Google Scholar] [CrossRef] [PubMed]
- Vilcheze, C.; Av-Gay, Y.; Attarian, R.; Liu, Z.; Hazbon, M.H.; Colangeli, R.; Chen, B.; Liu, W.; Alland, D.; Sacchettini, J.C.; et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2008, 69, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Vilcheze, C.; Weisbrod, T.R.; Chen, B.; Kremer, L.; Hazbon, M.H.; Wang, F.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R., Jr. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob. Agents Chemother. 2005, 49, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Baulard, A.R.; Betts, J.C.; Engohang-Ndong, J.; Quan, S.; McAdam, R.A.; Brennan, P.J.; Locht, C.; Besra, G.S. Activation of the pro-drug ethionamide is regulated in mycobacteria. J. Biol. Chem. 2000, 275, 28326–28331. [Google Scholar] [CrossRef] [PubMed]
- Willand, N.; Dirie, B.; Carette, X.; Bifani, P.; Singhal, A.; Desroses, M.; Leroux, F.; Willery, E.; Mathys, V.; Deprez-Poulain, R.; et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 2009, 15, 537–544. [Google Scholar] [CrossRef]
- Flipo, M.; Desroses, M.; Lecat-Guillet, N.; Villemagne, B.; Blondiaux, N.; Leroux, F.; Piveteau, C.; Mathys, V.; Flament, M.P.; Siepmann, J.; et al. Ethionamide boosters. 2. Combining bioisosteric replacement and structure-based drug design to solve pharmacokinetic issues in a series of potent 1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 2012, 55, 68–83. [Google Scholar] [CrossRef]
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 2017, 355, 1206–1211. [Google Scholar] [CrossRef]
- Harris, D.A.; Ruger, M.; Reagan, M.A.; Wolf, F.J.; Peck, R.L.; Wallick, H.; Woodruff, H.B. Discovery, development, and antimicrobial properties of D-4-amino-3-isoxazolidone (oxamycin), a new antibiotic produced by Streptomyces garyphalus n. sp. Antibiot. Chemother. 1955, 5, 183–190. [Google Scholar]
- Shull, G.M.; Sardinas, J.L. PA-94, an antibiotic identical with D-4-amino-3-isoxazolidinone (cycloserine, oxamycin). Antibiot. Chemother. 1955, 5, 398–399. [Google Scholar]
- Kurosawa, H. The isolation of an antibiotic produced by a strain of streptomyces K-300. Yokohama Med. Bull. 1952, 3, 386–399. [Google Scholar]
- Harned, R.L.; Hidy, P.H.; La Baw, E.K. Cycloserine. I. A preliminary report. Antibiot. Chemother. 1955, 5, 204–205. [Google Scholar]
- Steenken, W., Jr.; Wolinsky, E. Cycloserine: Antituberculous activity in vitro and in the experimental animal. Am. Rev. Tuberc. 1956, 73, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Cuckler, A.C.; Frost, B.M.; Mc, C.L.; Solotorovsky, M. The antimicrobial evaluation of oxamycin (D-4-amino-3-isoxazolidone), a new broad-spectrum antibiotic. Antibiot. Chemother. 1955, 5, 191–197. [Google Scholar]
- Patnode, R.A.; Hudgins, P.C.; Cummings, M.M. Effect of cycloserine on experimental tuberculosis in guinea pigs. Am. Rev. Tuberc. 1955, 72, 117–118. [Google Scholar] [CrossRef]
- Conzelman, G.M., Jr.; Jones, R.K. On the physiologic disposition of cycloserine in experimental animals. Am. Rev. Tuberc. 1956, 74, 802–806. [Google Scholar] [CrossRef]
- Epstein, I.G.; Nair, K.G.; Boyd, L.J. Cycloserine, a new antibiotic, in the treatment of human pulmonary tuberculosis: A preliminary report. Antibiot. Med. Clin. Ther. 1955, 1, 80–93. [Google Scholar]
- Lester, W., Jr.; Salomon, A.; Reimann, A.F.; Shulruff, E.; Berg, G.S. Cycloserine therapy in tuberculosis in humans. Am. Rev. Tuberc. 1956, 74, 121–127. [Google Scholar] [CrossRef]
- Hoeprich, P.D. Alanine: Cycloserine Antagonism. Vi. Demonstration of D-Alanine in the Serum of Guinea Pigs and Mice. J. Biol. Chem. 1965, 240, 1654–1660. [Google Scholar]
- Li, Y.; Wang, F.; Wu, L.; Zhu, M.; He, G.; Chen, X.; Sun, F.; Liu, Q.; Wang, X.; Zhang, W. Cycloserine for treatment of multidrug-resistant tuberculosis: A retrospective cohort study in China. Infect. Drug Resist. 2019, 12, 721–731. [Google Scholar] [CrossRef]
- Walker, W.C.; Murdoch, J.M. Cycloserine in the treatment of pulmonary tuberculosis; a report on toxicity. Tubercle 1957, 38, 297–302. [Google Scholar] [CrossRef]
- Bondi, A.; Kornblum, J.; Forte, C. Inhibition of antibacterial activity of cycloserine by alpha-alanine. Proc. Soc. Exp. Biol. Med. 1957, 96, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Strominger, J.L.; Ito, E.; Threnn, R.H. Competitive inhibition of enzymatic reactions by oxamycin. J. Am. Chem. Soc. 1960, 82, 998–999. [Google Scholar] [CrossRef]
- Halouska, S.; Fenton, R.J.; Zinniel, D.K.; Marshall, D.D.; Barletta, R.G.; Powers, R. Metabolomics analysis identifies d-Alanine-d-Alanine ligase as the primary lethal target of d-Cycloserine in mycobacteria. J. Proteome Res. 2014, 13, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Prosser, G.A.; de Carvalho, L.P. Reinterpreting the mechanism of inhibition of Mycobacterium tuberculosis D-alanine:D-alanine ligase by D-cycloserine. Biochemistry 2013, 52, 7145–7149. [Google Scholar] [CrossRef]
- Prosser, G.A.; de Carvalho, L.P. Kinetic mechanism and inhibition of Mycobacterium tuberculosis D-alanine:D-alanine ligase by the antibiotic D-cycloserine. FEBS J. 2013, 280, 1150–1166. [Google Scholar] [CrossRef]
- Viallier, J.; Cayre, R.M.; Biot, N. Sensitivity and resistance of Mycobacterium tuberculosis to cycloserine; study of 115 strains isolated from human pathological materials. Ann. Inst. Pasteur (Paris) 1957, 93, 127–131. [Google Scholar]
- Chen, J.M.; Uplekar, S.; Gordon, S.V.; Cole, S.T. A point mutation in cycA partially contributes to the D-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS ONE 2012, 7, e43467. [Google Scholar] [CrossRef]
- Desjardins, C.A.; Cohen, K.A.; Munsamy, V.; Abeel, T.; Maharaj, K.; Walker, B.J.; Shea, T.P.; Almeida, D.V.; Manson, A.L.; Salazar, A.; et al. Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat. Genet. 2016, 48, 544–551. [Google Scholar] [CrossRef]
- Nakatani, Y.; Opel-Reading, H.K.; Merker, M.; Machado, D.; Andres, S.; Kumar, S.S.; Moradigaravand, D.; Coll, F.; Perdigao, J.; Portugal, I.; et al. Role of Alanine Racemase Mutations in Mycobacterium tuberculosis d-Cycloserine Resistance. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Cui, P.; Shi, W.; Zhang, W.; Zhang, Y. Identification of novel mutations associated with cycloserine resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2017, 72, 3272–3276. [Google Scholar] [CrossRef]
- Evangelopoulos, D.; Prosser, G.A.; Rodgers, A.; Dagg, B.M.; Khatri, B.; Ho, M.M.; Gutierrez, M.G.; Cortes, T.; de Carvalho, L.P.S. Comparative fitness analysis of D-cycloserine resistant mutants reveals both fitness-neutral and high-fitness cost genotypes. Nat. Commun. 2019, 10, 4177. [Google Scholar] [CrossRef] [PubMed]
- Hager, T. The Demon under the Microscope. From Battlefield Hospitals to Nazi Labs, One Doctor’s Heroic Search for the World’s First Miracle Drug, 1st ed.; Three River Press: New York, NY, USA, 2006; p. 340. [Google Scholar]
- Domagk, G. Investigations on the antituberculous activity of the thiosemicarbazones in vitro and in vivo. Am. Rev. Tuberc. 1950, 61, 8–19. [Google Scholar] [PubMed]
- Mertens, A.; Bunge, R. The present status of the chemotherapy of tuberculosis with conteben a substance of the thiosemicarbazone series; a review. Am. Rev. Tuberc. 1950, 61, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, P.; McPherson, K. The thiacetazone sensitivity of Mycobacterium tuberculosis. J. Med. Microbiol. 1969, 2, 237–242. [Google Scholar] [CrossRef]
- Rieder, H.L.; Enarson, D.A. Rebuttal: Time to call a halt to emotions in the assessment of thioacetazone. Tuber. Lung Dis. 1996, 77, 109–111. [Google Scholar] [CrossRef]
- Falzon, D.; Hill, G.; Pal, S.N.; Suwankesawong, W.; Jaramillo, E. Pharmacovigilance and tuberculosis: Applying the lessons of thioacetazone. Bull. World Health Organ. 2014, 92, 918–919. [Google Scholar] [CrossRef]
- Dover, L.G.; Alahari, A.; Gratraud, P.; Gomes, J.M.; Bhowruth, V.; Reynolds, R.C.; Besra, G.S.; Kremer, L. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrob. Agents Chemother. 2007, 51, 1055–1063. [Google Scholar] [CrossRef]
- Belardinelli, J.M.; Morbidoni, H.R. Mutations in the essential FAS II beta-hydroxyacyl ACP dehydratase complex confer resistance to thiacetazone in Mycobacterium tuberculosis and Mycobacterium kansasii. Mol. Microbiol. 2012, 86, 568–579. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Kordulakova, J.; Jones, V.; Born, S.E.; Belardinelli, J.M.; Vaquie, A.; Gundi, V.A.; Madacki, J.; Slama, N.; Laval, F.; et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef]
- Dong, Y.; Qiu, X.; Shaw, N.; Xu, Y.; Sun, Y.; Li, X.; Li, J.; Rao, Z. Molecular basis for the inhibition of beta-hydroxyacyl-ACP dehydratase HadAB complex from Mycobacterium tuberculosis by flavonoid inhibitors. Protein Cell 2015, 6, 504–517. [Google Scholar] [CrossRef]
- Grzegorzewicz, A.E.; Eynard, N.; Quemard, A.; North, E.J.; Margolis, A.; Lindenberger, J.J.; Jones, V.; Kordulakova, J.; Brennan, P.J.; Lee, R.E.; et al. Covalent modification of the Mycobacterium tuberculosis FAS-II dehydratase by Isoxyl and Thiacetazone. ACS Infect. Dis. 2015, 1, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Coxon, G.D.; Craig, D.; Corrales, R.M.; Vialla, E.; Gannoun-Zaki, L.; Kremer, L. Synthesis, antitubercular activity and mechanism of resistance of highly effective thiacetazone analogues. PLoS ONE 2013, 8, e53162. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewicz, A.E.; Gee, C.; Das, S.; Liu, J.; Belardinelli, J.M.; Jones, V.; McNeil, M.R.; Lee, R.E.; Jackson, M. Mechanisms of Resistance Associated with the Inhibition of the Dehydration Step of Type II Fatty Acid Synthase in Mycobacterium tuberculosis. ACS Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Alahari, A.; Alibaud, L.; Trivelli, X.; Gupta, R.; Lamichhane, G.; Reynolds, R.C.; Bishai, W.R.; Guerardel, Y.; Kremer, L. Mycolic acid methyltransferase, MmaA4, is necessary for thiacetazone susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2009, 71, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E., 3rd; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Alland, D.; Steyn, A.J.; Weisbrod, T.; Aldrich, K.; Jacobs, W.R., Jr. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 2000, 182, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef]
- Chen, P.; Gearhart, J.; Protopopova, M.; Einck, L.; Nacy, C.A. Synergistic interactions of SQ109, a new ethylene diamine, with front-line antitubercular drugs in vitro. J. Antimicrob. Chemother. 2006, 58, 332–337. [Google Scholar] [CrossRef]
- Jia, L.; Tomaszewski, J.E.; Hanrahan, C.; Coward, L.; Noker, P.; Gorman, G.; Nikonenko, B.; Protopopova, M. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005, 144, 80–87. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Myers, T.G.; Copp, B.R.; McNeil, M.R.; Wilson, M.A.; Barry, C.E., 3rd. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: Novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 279, 40174–40184. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., 3rd; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.; Kordulakova, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Cheng, X.; Liu, Z.; et al. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648.e13. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivisto, K.T. Pharmacokinetic interactions with rifampicin: Clinical relevance. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef]
- Boeree, M.J.; Heinrich, N.; Aarnoutse, R.; Diacon, A.H.; Dawson, R.; Rehal, S.; Kibiki, G.S.; Churchyard, G.; Sanne, I.; Ntinginya, N.E.; et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: A multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis. 2017, 17, 39–49. [Google Scholar] [CrossRef]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198. [Google Scholar] [CrossRef]
- Poce, G.; Consalvi, S.; Biava, M. MmpL3 Inhibitors: Diverse Chemical Scaffolds Inhibit the Same Target. Mini Rev. Med. Chem. 2016, 16, 1274–1283. [Google Scholar] [CrossRef]
- Williams, J.T.; Haiderer, E.R.; Coulson, G.B.; Conner, K.N.; Ellsworth, E.; Chen, C.; Alvarez-Cabrera, N.; Li, W.; Jackson, M.; Dick, T.; et al. Identification of New MmpL3 Inhibitors by Untargeted and Targeted Mutant Screens Defines MmpL3 Domains with Differential Resistance. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Samuelson, J. Why metronidazole is active against both bacteria and parasites. Antimicrob. Agents Chemother. 1999, 43, 1533–1541. [Google Scholar] [CrossRef]
- Brooks, J.V.; Furney, S.K.; Orme, I.M. Metronidazole therapy in mice infected with tuberculosis. Antimicrob. Agents Chemother. 1999, 43, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Ashtekar, D.R.; Costa-Perira, R.; Nagrajan, K.; Vishvanathan, N.; Bhatt, A.D.; Rittel, W. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993, 37, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and Sterilizing Activity of a Novel Regimen with Bedaquiline, Pretomanid, Moxifloxacin, and Pyrazinamide in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, S.Y.; Almeida, D.V.; Tasneen, R.; Barnes-Boyle, K.; Converse, P.J.; Upton, A.M.; Mdluli, K.; Fotouhi, N.; Nuermberger, E.L. Contribution of Pretomanid to Novel Regimens Containing Bedaquiline with either Linezolid or Moxifloxacin and Pyrazinamide in Murine Models of Tuberculosis. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Manjunatha, U.; Boshoff, H.I.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 2008, 322, 1392–1395. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef]
- Haver, H.L.; Chua, A.; Ghode, P.; Lakshminarayana, S.B.; Singhal, A.; Mathema, B.; Wintjens, R.; Bifani, P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 5316–5323. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; von Groote-Bidlingmaier, F.; Symons, G.; Venter, A.; Donald, P.R.; van Niekerk, C.; Everitt, D.; Winter, H.; Becker, P.; et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: A randomised trial. Lancet 2012, 380, 986–993. [Google Scholar] [CrossRef]
- Tweed, C.D.; Dawson, R.; Burger, D.A.; Conradie, A.; Crook, A.M.; Mendel, C.M.; Conradie, F.; Diacon, A.H.; Ntinginya, N.E.; Everitt, D.E.; et al. Bedaquiline, moxifloxacin, pretomanid, and pyrazinamide during the first 8 weeks of treatment of patients with drug-susceptible or drug-resistant pulmonary tuberculosis: A multicentre, open-label, partially randomised, phase 2b trial. Lancet Respir. Med. 2019, 7, 1048–1058. [Google Scholar] [CrossRef]
- Conradie, F.; Diacon, A.; Everitt, D.; Mendel, C.M.; Crook, A.M.; Howell, P.; Comins, K.; Spigelman, M. Sustained High Rate of Successful Treatment Outcomes: Interim Results of 75 Patients in the Nix-TB Clinical Study of Pretomanid, Bedaquiline and Linezolid. Available online: https://www.dropbox.com/s/gu8l27grq38psul/Nix%20TB%20interim%20results%20-%2010-25-18.pdf?dl=0 (accessed on 30 January 2020).
- Sasaki, H.; Haraguchi, Y.; Itotani, M.; Kuroda, H.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Matsumoto, M.; Komatsu, M.; Tsubouchi, H. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J. Med. Chem. 2006, 49, 7854–7860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Matsumoto, M.; Ishida, H.; Ohguro, K.; Yoshitake, M.; Gupta, R.; Geiter, L.; Hafkin, J. Delamanid: From discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef] [PubMed]
- Upton, A.M.; Cho, S.; Yang, T.J.; Kim, Y.; Wang, Y.; Lu, Y.; Wang, B.; Xu, J.; Mdluli, K.; Ma, Z.; et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Kawasaki, M.; Hariguchi, N.; Liu, Y.; Matsumoto, M. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis 2018, 108, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Gler, M.T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J.L.; Vargas-Vasquez, D.E.; Gao, M.; Awad, M.; Park, S.K.; Shim, T.S.; et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. [Google Scholar] [CrossRef]
- Gupta, R.; Geiter, L.J.; Wells, C.D.; Gao, M.; Cirule, A.; Xiao, H. Delamanid for Extensively Drug-Resistant Tuberculosis. N. Engl. J. Med. 2015, 373, 291–292. [Google Scholar] [CrossRef]
- Wells, C.D.; Gupta, R.; Hittel, N.; Geiter, L.J. Long-term mortality assessment of multidrug-resistant tuberculosis patients treated with delamanid. Eur. Respir. J. 2015, 45, 1498–1501. [Google Scholar] [CrossRef]
- Von Groote-Bidlingmaier, F.; Patientia, R.; Sanchez, E.; Balanag, V., Jr.; Ticona, E.; Segura, P.; Cadena, E.; Yu, C.; Cirule, A.; Lizarbe, V.; et al. Efficacy and safety of delamanid in combination with an optimised background regimen for treatment of multidrug-resistant tuberculosis: A multicentre, randomised, double-blind, placebo-controlled, parallel group phase 3 trial. Lancet Respir. Med. 2019, 7, 249–259. [Google Scholar] [CrossRef]
- Kumar, P.; Capodagli, G.C.; Awasthi, D.; Shrestha, R.; Maharaja, K.; Sukheja, P.; Li, S.G.; Inoyama, D.; Zimmerman, M.; Ho Liang, H.P.; et al. Synergistic Lethality of a Binary Inhibitor of Mycobacterium tuberculosis KasA. MBio 2018, 9. [Google Scholar] [CrossRef]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259.e25. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, M.; Selchow, P.; Schafle, D.; Tschumi, A.; Ryckmans, T.; Laage-Witt, S.; Sander, P. Identification of novel scaffolds targeting Mycobacterium tuberculosis. J. Mol. Med. 2019, 97, 1601–1613. [Google Scholar] [CrossRef]
- Wilson, R.; Kumar, P.; Parashar, V.; Vilcheze, C.; Veyron-Churlet, R.; Freundlich, J.S.; Barnes, S.W.; Walker, J.R.; Szymonifka, M.J.; Marchiano, E.; et al. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 2013, 9, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lun, S.; Wang, S.H.; Jiang, X.W.; Yang, F.; Tang, J.; Manson, A.L.; Earl, A.M.; Gunosewoyo, H.; Bishai, W.R.; et al. Identification of Novel Coumestan Derivatives as Polyketide Synthase 13 Inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sanchez-Hidalgo, A.; Jones, V.; de Moura, V.C.; North, E.J.; Jackson, M. Synergistic Interactions of MmpL3 Inhibitors with Antitubercular Compounds In Vitro. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Poce, G.; Bates, R.H.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Ballell, L.; Rullas, J.; Ortega, F.; De Logu, A.; Agus, E.; et al. Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection. PLoS ONE 2013, 8, e56980. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Nakagawa, N.; Doi, N.; Hattori, S.; Naganawa, H.; Hamada, M. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J. Antibiot. 2003, 56, 580–583. [Google Scholar] [CrossRef]
- Takahashi, Y.; Igarashi, M.; Miyake, T.; Soutome, H.; Ishikawa, K.; Komatsuki, Y.; Koyama, Y.; Nakagawa, N.; Hattori, S.; Inoue, K.; et al. Novel semisynthetic antibiotics from caprazamycins A-G: Caprazene derivatives and their antibacterial activity. J. Antibiot. 2013, 66, 171–178. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Hayashi, C.; Inoue, K.; Igarashi, M.; Takahashi, Y.; Pujari, V.; Crick, D.C.; Brennan, P.J.; Nomoto, A. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J. Biol. Chem. 2013, 288, 30309–30319. [Google Scholar] [CrossRef]
- Takahashi, Y.; Igarashi, M.; Okada, M. Anti-XDR-TB, Anti-MDR-TB Drug, and Combination Anti-Tuberculoses Drug. U.S. Patent 9040502 B2, 26 May 2015. [Google Scholar]
- Huszar, S.; Singh, V.; Polcicova, A.; Barath, P.; Barrio, M.B.; Lagrange, S.; Leblanc, V.; Nacy, C.A.; Mizrahi, V.; Mikusova, K. N-Acetylglucosamine-1-Phosphate Transferase, WecA, as a Validated Drug Target in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Mollmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Lechartier, B.; Hartkoorn, R.C.; Cole, S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5790–5793. [Google Scholar] [CrossRef]
- New TB Drugs. BTZ-043. Available online: https://www.newtbdrugs.org/pipeline/compound/btz-043 (accessed on 15 February 2020).
- Trefzer, C.; Skovierova, H.; Buroni, S.; Bobovska, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T.; et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-beta-D-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Lupien, A.; Vocat, A.; Foo, C.S.; Blattes, E.; Gillon, J.Y.; Makarov, V.; Cole, S.T. Optimized Background Regimen for Treatment of Active Tuberculosis with the Next-Generation Benzothiazinone Macozinone (PBTZ169). Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Lechartier, B.; Cole, S.T. Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Workshop “Critical Path to TB Drug Regimens”. Available online: http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf (accessed on 26 January 2020).
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-Resistance between Clofazimine and Bedaquiline through Upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Prabhakar, K.R.; et al. 1,4-azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Peng, C.; Shi, Y.; You, X.; Ran, K.; Xiong, L.; Ye, T.H.; Zhang, L.; Wang, N.; Zhu, Y.; et al. Benzothiazinethione is a potent preclinical candidate for the treatment of drug-resistant tuberculosis. Sci. Rep. 2016, 6, 29717. [Google Scholar] [CrossRef] [PubMed]
- Christophe, T.; Jackson, M.; Jeon, H.K.; Fenistein, D.; Contreras-Dominguez, M.; Kim, J.; Genovesio, A.; Carralot, J.P.; Ewann, F.; Kim, E.H.; et al. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog. 2009, 5, e1000645. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Hartkoorn, R.C.; Szekely, R.; Pato, J.; Triccas, J.A.; Schneider, P.; Szantai-Kis, C.; Orfi, L.; Chambon, M.; Banfi, D.; et al. Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 2010, 90, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Grant, S.S.; Kawate, T.; Iwase, N.; Shimizu, M.; Wivagg, C.; Silvis, M.; Kazyanskaya, E.; Aquadro, J.; Golas, A.; et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 2012, 7, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E2510–E2517. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-aminoquinolone piperidine amides: Noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines kill Mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef]
- Makarov, V.; Neres, J.; Hartkoorn, R.C.; Ryabova, O.B.; Kazakova, E.; Sarkan, M.; Huszar, S.; Piton, J.; Kolly, G.S.; Vocat, A.; et al. The 8-Pyrrole-Benzothiazinones Are Noncovalent Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4446–4452. [Google Scholar] [CrossRef]
- Warrier, T.; Kapilashrami, K.; Argyrou, A.; Ioerger, T.R.; Little, D.; Murphy, K.C.; Nandakumar, M.; Park, S.; Gold, B.; Mi, J.; et al. N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4523–E4530. [Google Scholar] [CrossRef]
- Piton, J.; Vocat, A.; Lupien, A.; Foo, C.S.; Riabova, O.; Makarov, V.; Cole, S.T. Structure-Based Drug Design and Characterization of Sulfonyl-Piperazine Benzothiazinone Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Borthwick, J.A.; Alemparte, C.; Wall, I.; Whitehurst, B.C.; Argyrou, A.; Burley, G.; de Dios-Anton, P.; Guijarro, L.; Monteiro, M.C.; Ortega, F.; et al. Mycobacterium tuberculosis Decaprenylphosphoryl-beta-d-ribose Oxidase Inhibitors: Expeditious Reconstruction of Suboptimal Hits into a Series with Potent in Vivo Activity. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hugonnet, J.E.; Blanchard, J.S. Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 2007, 46, 11998–12004. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.R.; Parsons, L.M.; Pavelka, M.S. Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 2005, 151, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Hugonnet, J.E.; Tremblay, L.W.; Boshoff, H.I.; Barry, C.E., 3rd; Blanchard, J.S. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 2009, 323, 1215–1218. [Google Scholar] [CrossRef]
- England, K.; Boshoff, H.I.; Arora, K.; Weiner, D.; Dayao, E.; Schimel, D.; Via, L.E.; Barry, C.E., 3rd. Meropenem-clavulanic acid shows activity against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 2012, 56, 3384–3387. [Google Scholar] [CrossRef]
- Veziris, N.; Truffot, C.; Mainardi, J.L.; Jarlier, V. Activity of carbapenems combined with clavulanate against murine tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2597–2600. [Google Scholar] [CrossRef]
- Dhar, N.; Dubee, V.; Ballell, L.; Cuinet, G.; Hugonnet, J.E.; Signorino-Gelo, F.; Barros, D.; Arthur, M.; McKinney, J.D. Rapid cytolysis of Mycobacterium tuberculosis by faropenem, an orally bioavailable beta-lactam antibiotic. Antimicrob. Agents Chemother. 2014, 59, 1308–1319. [Google Scholar] [CrossRef]
- Rullas, J.; Dhar, N.; McKinney, J.D.; Garcia-Perez, A.; Lelievre, J.; Diacon, A.H.; Hugonnet, J.E.; Arthur, M.; Angulo-Barturen, I.; Barros-Aguirre, D.; et al. Combinations of beta-Lactam Antibiotics Currently in Clinical Trials Are Efficacious in a DHP-I-Deficient Mouse Model of Tuberculosis Infection. Antimicrob. Agents Chemother. 2015, 59, 4997–4999. [Google Scholar] [CrossRef]
- Dubee, V.; Triboulet, S.; Mainardi, J.L.; Etheve-Quelquejeu, M.; Gutmann, L.; Marie, A.; Dubost, L.; Hugonnet, J.E.; Arthur, M. Inactivation of Mycobacterium tuberculosis L,D-transpeptidase LdtMt1 by carbapenems and cephalosporins. Antimicrob. Agents Chemother. 2012, 56, 4189–4195. [Google Scholar] [CrossRef]
- Lavollay, M.; Arthur, M.; Fourgeaud, M.; Dubost, L.; Marie, A.; Veziris, N.; Blanot, D.; Gutmann, L.; Mainardi, J.L. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J. Bacteriol. 2008, 190, 4360–4366. [Google Scholar] [CrossRef]
- Tiberi, S.; Payen, M.C.; Sotgiu, G.; D’Ambrosio, L.; Alarcon Guizado, V.; Alffenaar, J.W.; Abdo Arbex, M.; Caminero, J.A.; Centis, R.; De Lorenzo, S.; et al. Effectiveness and safety of meropenem/clavulanate-containing regimens in the treatment of MDR- and XDR-TB. Eur. Respir. J. 2016, 47, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Di Modugno, E.; Erbetti, I.; Ferrari, L.; Galassi, G.; Hammond, S.M.; Xerri, L. In vitro activity of the tribactam GV104326 against gram-positive, gram-negative, and anaerobic bacteria. Antimicrob. Agents Chemother. 1994, 38, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Barros Aguirre, D.; Bates, R.H.; Gonzalez Del Rio, R.; Mendoza Losana, A.; Ramón García, S. Sanfetrinem or a Salt or Ester Thereof for Use in Treating Mycobacterial Infection. Patent No. WO/2018/206466, 15 November 2018. [Google Scholar]
- New TB Drugs. Sanfetrinem. Available online: https://www.newtbdrugs.org/pipeline/compound/sanfetrinem (accessed on 31 January 2020).
- Campanico, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Libardo, M.; Boshoff, H.I.; Barry, C.E., 3rd. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; Munoz-Torrico, M.; Duarte, R.; Dalcolmo, M.; D’Ambrosio, L.; Migliori, G.B. New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef]
- Waksman, S.A. The Conquest of Tuberculosis; University of California Press: Berkeley, CA, USA, 1964; p. 211. [Google Scholar]



{kind=link}
{kind=link}
| Name | Structure | Cell Wall Component Inhibited | Prodrug/Activator | Target |
|---|---|---|---|---|
| Isoniazid |  | Mycolic acids | + KatG | InhA |
| Ethionamide Prothionamide |  | Mycolic acids | + EthA, MymA, Rv0565c | InhA |
| Thiacetazone |  | Mycolic acids | + EthA | HadAB |
| Pretonamid |  | Keto-mycolic acids | + Ddn | ? |
| Delamanid |  | Methoxy- and keto-mycolic acids | + Ddn | ? |
| SQ109 |  | Mycolic acid transport | - | Mmpl3 |
| CPZEN45 |  | Arabinogalactan | - | WecA |
| Ethambutol |  | Arabinogalactan LAM | - | EmbCAB |
| BTZ043 |  | Arabinogalactan LAM | + DprE1 | DprE1 |
| PBTZ169 |  | Arabinogalactan LAM | + DprE1 | DprE1 |
| OPC167832 |  | Arabinogalactan LAM | - | DprE1 |
| TBA7371 |  | Arabinogalactan LAM | - | DprE1 |
| Cycloserine |  | Peptidoglycan | - | DdlA |
| Meropenem/clavulanate |  | Peptidoglycan | - | LdtM1 |
| sanfetrinem |  | Peptidoglycan | - |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 2020, 10, 2278. https://doi.org/10.3390/app10072278
Vilchèze C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Applied Sciences. 2020; 10(7):2278. https://doi.org/10.3390/app10072278
Chicago/Turabian StyleVilchèze, Catherine. 2020. "Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs" Applied Sciences 10, no. 7: 2278. https://doi.org/10.3390/app10072278
APA StyleVilchèze, C. (2020). Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Applied Sciences, 10(7), 2278. https://doi.org/10.3390/app10072278