URSA-PQ: A Mobile and Flexible Pump-Probe Instrument for Gas Phase Samples at the FLASH Free Electron Laser

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
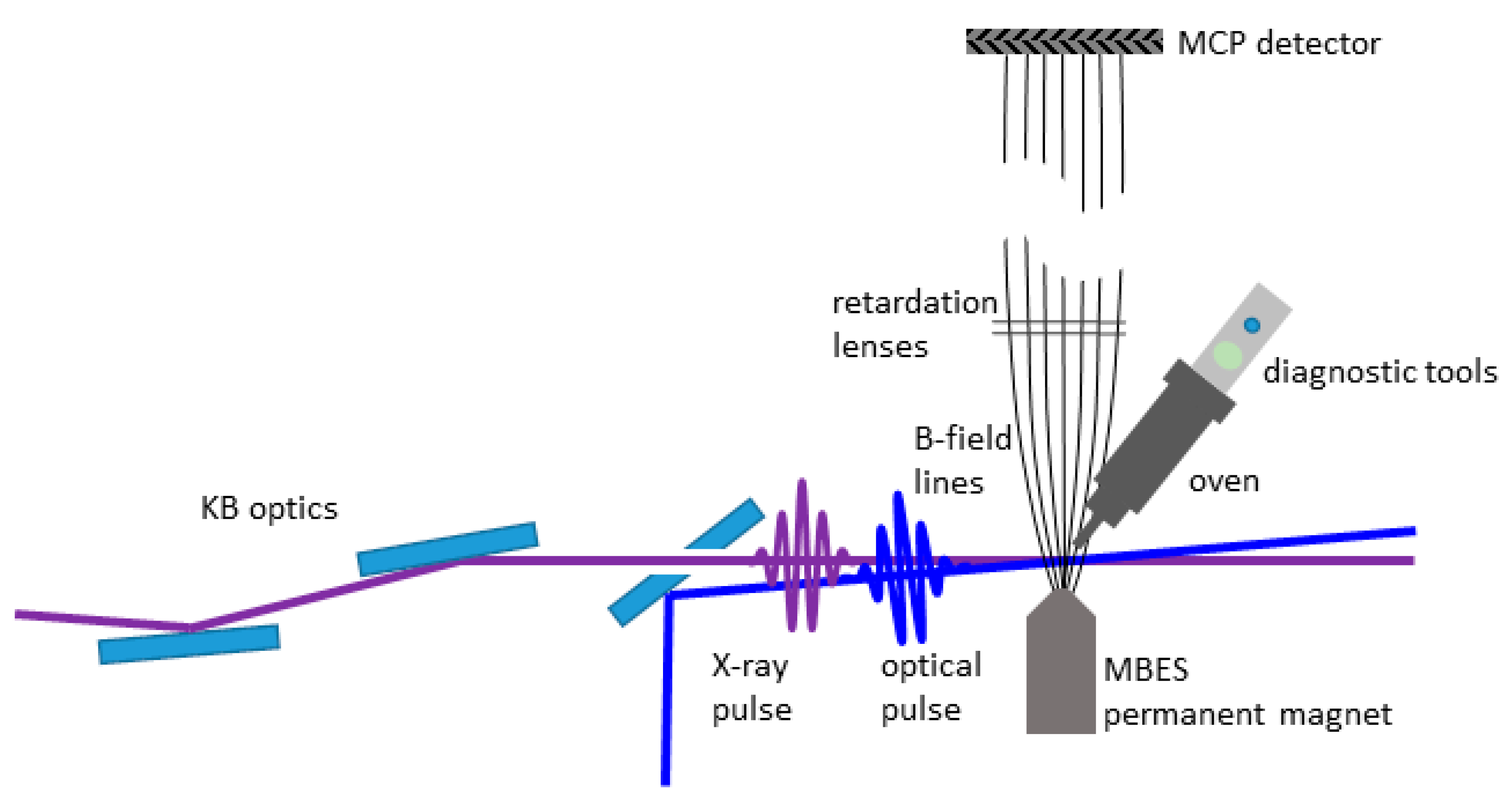
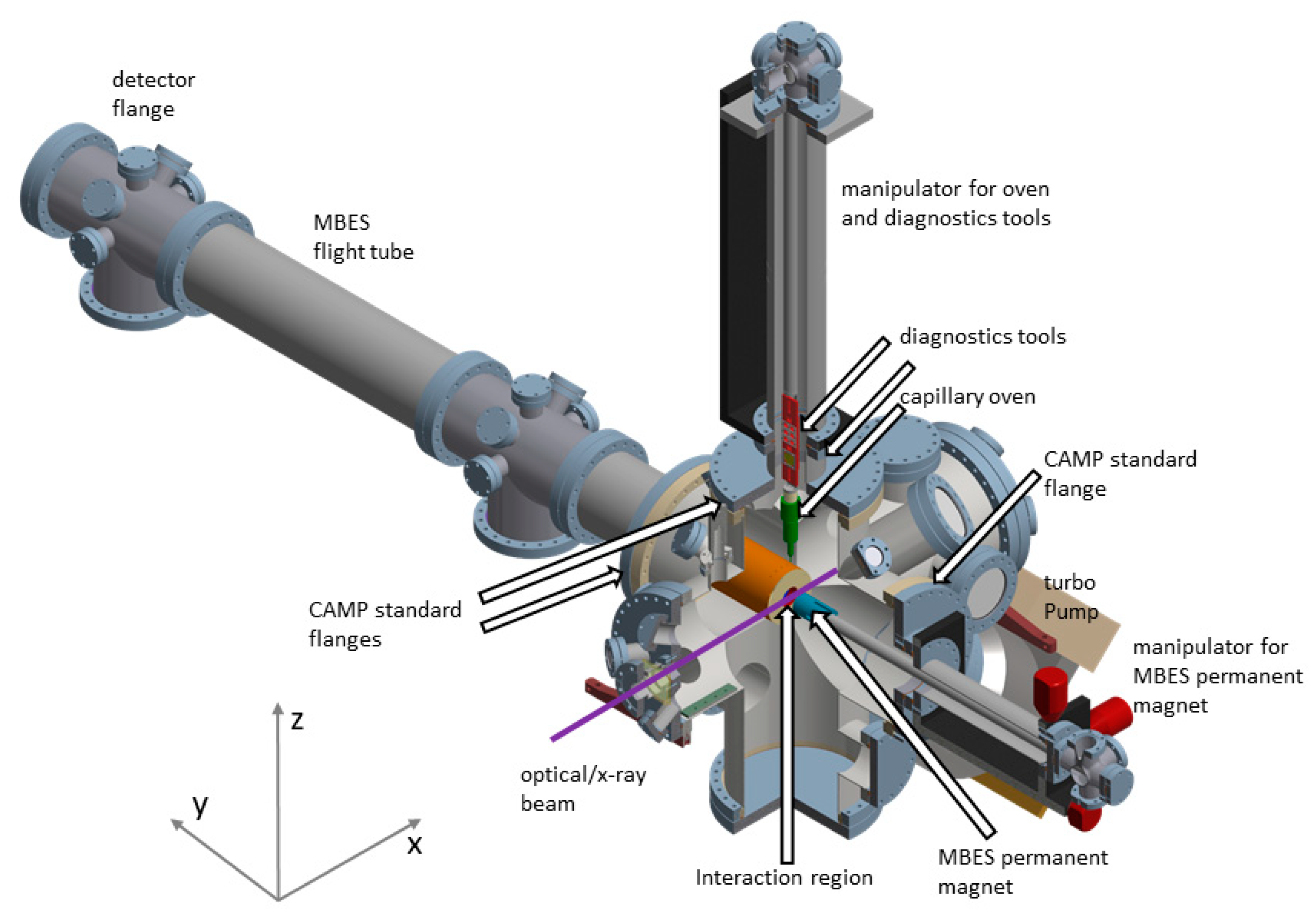
2.1. Overview and Vacuum System
2.2. Integration at the FLASH FL 24 Beamline
2.3. Diagnostics and Oven
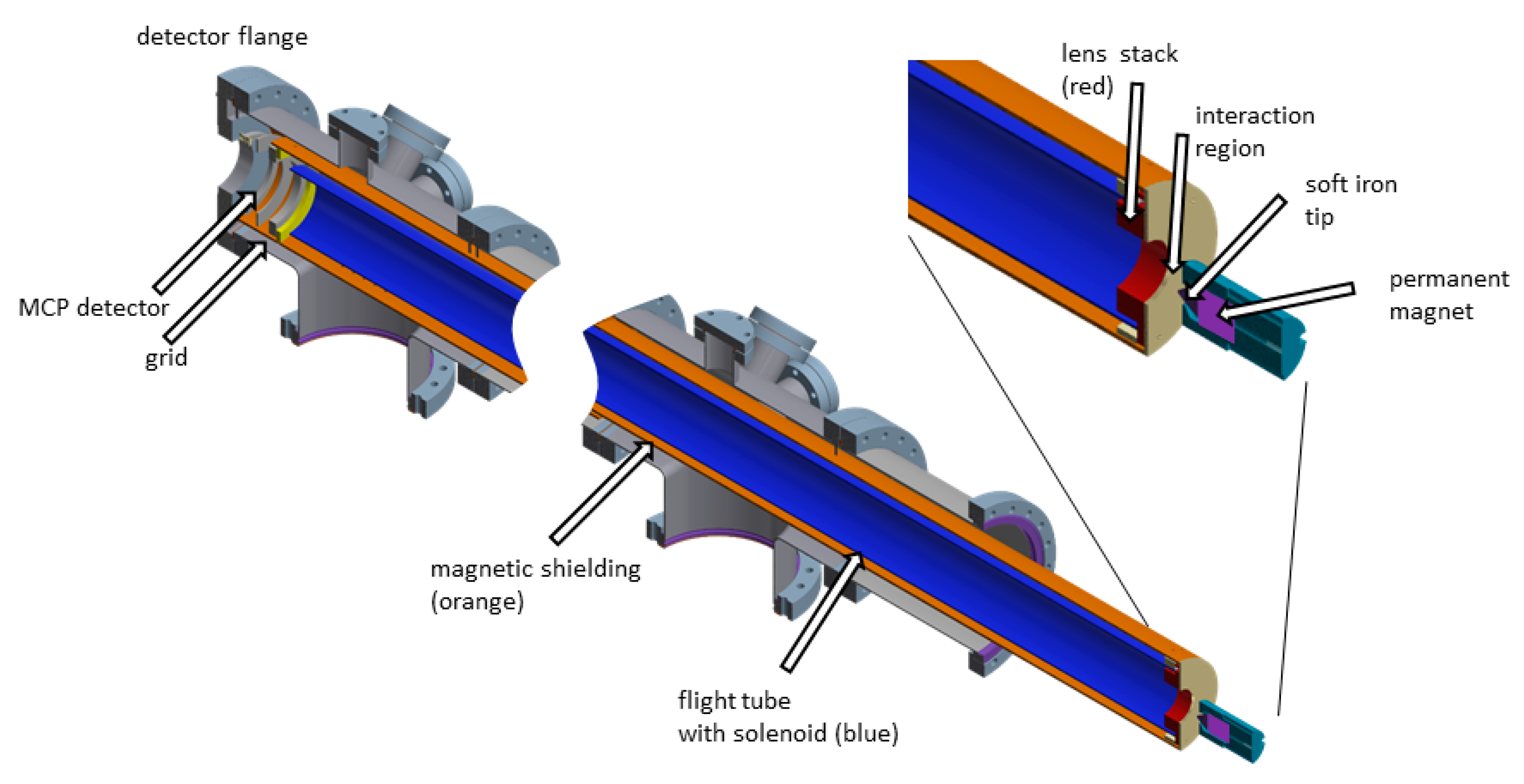
2.4. Magnetic Bottle Electron Spectrometer
3. Results and Discussion
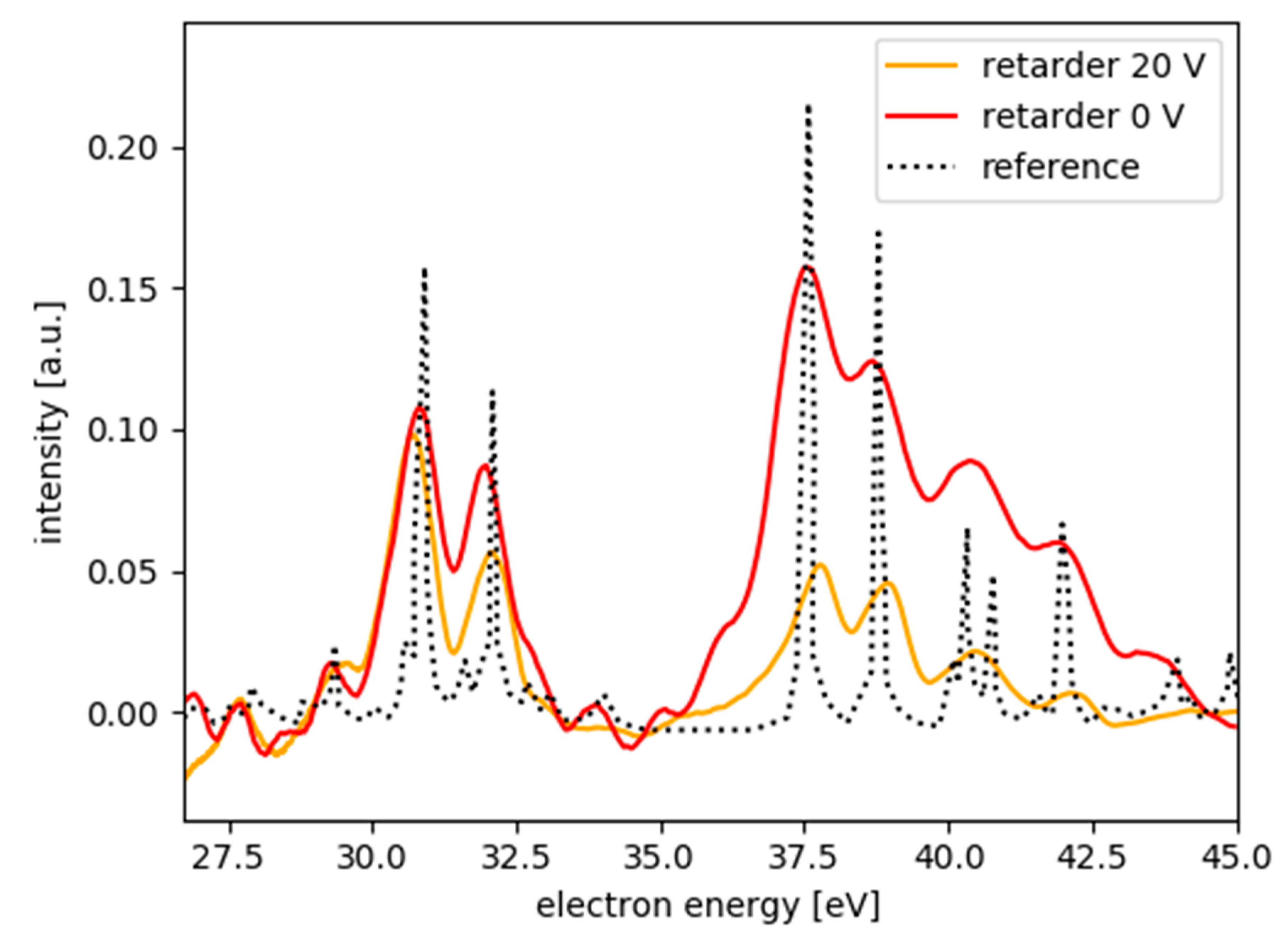
3.1. MBES Energy Resolution
3.2. Temporal Resolution
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yarkony, D. Diabolical conical intersections. Rev. Mod. Phys. 1996, 68, 985–1013. [Google Scholar] [CrossRef]
- Levine, B.G.; Martínez, T.J. Isomerization through Conical Intersections. Annu. Rev. Phys. Chem. 2007, 58, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Matsika, S.; Krause, P. Nonadiabatic Events and Conical Intersections. Annu. Rev. Phys. Chem. 2011, 62, 621–643. [Google Scholar] [CrossRef] [PubMed]
- Domcke, W.; Yarkony, D.R.; Köppel, H. Conical Intersections Electronic Structure, Dynamics and Spectroscopy; World Scientific Publishing Company: Singapore, 2011; ISBN 978-981-4313-45-2. [Google Scholar]
- Polli, D.; Altoè, P.; Weingart, O.; Spillane, K.M.; Manzoni, C.; Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R.A.; et al. Conical intersection dynamics of the primary photoisomerization event in vision. Nature 2010, 467, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast Excited-State Dynamics in Nucleic Acids. Chem. Rev. 2004, 104, 1977–2020. [Google Scholar] [CrossRef]
- Middleton, C.T.; de La Harpe, K.; Su, C.; Law, Y.K.; Crespo-Hernández, C.E.; Kohler, B. DNA Excited-State Dynamics: From Single Bases to the Double Helix. Annu. Rev. Phys. Chem. 2009, 60, 217–239. [Google Scholar] [CrossRef]
- Schreier, W.J.; Gilch, P.; Zinth, W. Early Events of DNA Photodamage. Annu. Rev. Phys. Chem. 2015, 66, 497–519. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef]
- Dörner, R.; Mergel, V.; Jagutzki, O.; Spielberger, L.; Ullrich, J.; Moshammer, R.; Schmidt-Böcking, H. Cold Target Recoil Ion Momentum Spectroscopy: A “momentum microscope” to view atomic collision dynamics. Phys. Rep. 2000, 330, 95–192. [Google Scholar] [CrossRef]
- Ullrich, J.; Moshammer, R.; Dorn, A.; Dorner, R.; Schmidt, L.P.H.; Schmidt-Böcking, H. Recoil-ion and electron momentum spectroscopy: Reaction-microscopes. Rep. Prog. Phys. 2003, 66, 1463–1545. [Google Scholar] [CrossRef]
- Stapelfeldt, H.; Seideman, T. Colloquium: Aligning molecules with strong laser pulses. Rev. Mod. Phys. 2003, 75, 543. [Google Scholar] [CrossRef]
- McFarland, B.K.; Farrell, J.P.; Bucksbaum, P.H.; Gühr, M. High Harmonic Generation from Multiple Orbitals in N2. Science 2008, 322, 1232. [Google Scholar] [CrossRef]
- Filsinger, F.; Meijer, G.; Stapelfeldt, H.; Chapman, H.N.; Kuepper, J. State- and conformer-selected beams of aligned and oriented molecules for ultrafast diffraction studies. Phys. Chem. Chem. Phys. 2011, 13, 2076–2087. [Google Scholar] [CrossRef]
- Siegbahn, K. ESCA Applied to Free Molecules; North-Holland Pub. Co.: Amsterdam, The Netherlands, 1969; ISBN 0-7204-0160-7. [Google Scholar]
- Wolf, T.J.A.; Myhre, R.H.; Cryan, J.P.; Coriani, S.; Squibb, R.J.; Battistoni, A.; Berrah, N.; Bostedt, C.; Bucksbaum, P.; Coslovich, G.; et al. Probing ultrafast ππ*/nπ* internal conversion in organic chromophores via K-edge resonant absorption. Nat. Commun. 2017, 8, 29. [Google Scholar] [CrossRef]
- Ehlert, C.; Gühr, M.; Saalfrank, P. An efficient first principles method for molecular pump-probe NEXAFS spectra: Application to thymine and azobenzene. J. Chem. Phys. 2018, 149, 144112. [Google Scholar] [CrossRef]
- McFarland, B.K.; Farrell, J.P.; Miyabe, S.; Tarantelli, F.; Aguilar, A.; Berrah, N.; Bostedt, C.; Bozek, J.D.; Bucksbaum, P.H.; Castagna, J.C.; et al. Ultrafast X-ray Auger probing of photoexcited molecular dynamics. Nat. Commun. 2014, 5, 4235. [Google Scholar] [CrossRef]
- Wolf, T.; Holzmeier, F.; Wagner, I.; Berrah, N.; Bostedt, C.; Bozek, J.; Bucksbaum, P.; Coffee, R.; Cryan, J.; Farrell, J.; et al. Observing Femtosecond Fragmentation Using Ultrafast X-ray-Induced Auger Spectra. Appl. Sci. 2017, 7, 681. [Google Scholar] [CrossRef]
- Kruit, P.; Read, F.H. Magnetic field paralleliser for 2π electron-spectrometer and electron-image magnifier. J. Phys. E Sci. Instrum. 1983, 16, 313–324. [Google Scholar] [CrossRef]
- McFarland, B.K.; Berrah, N.; Bostedt, C.; Bozek, J.; Bucksbaum, P.H.; Castagna, J.C.; Coffee, R.N.; Cryan, J.P.; Fang, L.; Farrell, J.P.; et al. Experimental strategies for optical pump – soft x-ray probe experiments at the LCLS. J. Phys. Conf. Ser. 2014, 488, 012015. [Google Scholar] [CrossRef]
- Ferro, D.; Bencivenni, L.; Teghil, R.; Mastromarino, R. Vapour pressures and sublimation enthalpies of thymine and cytosine. Thermochim. Acta 1980, 42, 75–83. [Google Scholar] [CrossRef]
- Strueder, L.; Epp, S.; Rolles, D.; Hartmann, R.; Holl, P.; Lutz, G.; Soltau, H.; Eckart, R.; Reich, C.; Heinzinger, K.; et al. Large-format, high-speed, X-ray pnCCDs combined with electron and ion imaging spectrometers in a multipurpose chamber for experiments at 4th generation light sources. Nucl. Instrum. Methods Phys. Res. Sect. A-Accel. Spectrom. Detect. Assoc. Equip. 2010, 614, 483–496. [Google Scholar] [CrossRef]
- Raimondi, L.; Manfredda, M.; Mahne, N.; Cocco, D.; Capotondi, F.; Pedersoli, E.; Kiskinova, M.; Zangrando, M. Kirkpatrick–Baez active optics system at FERMI: System performance analysis. J. Synchrotron Rad. 2019, 26, 1462–1472. [Google Scholar] [CrossRef]
- Zhaunerchyk, V.; Kamińska, M.; Mucke, M.; Squibb, R.J.; Eland, J.H.D.; Piancastelli, M.N.; Frasinski, L.J.; Grilj, J.; Koch, M.; McFarland, B.K.; et al. Disentangling formation of multiple-core holes in aminophenol molecules exposed to bright X-FEL radiation. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 244003. [Google Scholar] [CrossRef]
- Sanchez-Gonzalez, A.; Barillot, T.R.; Squibb, R.J.; Kolorenč, P.; Agaker, M.; Averbukh, V.; Bearpark, M.J.; Bostedt, C.; Bozek, J.D.; Bruce, S.; et al. Auger electron and photoabsorption spectra of glycine in the vicinity of the oxygen K-edge measured with an X-FEL. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 234004. [Google Scholar] [CrossRef]
- Koch, M.; Wolf, T.J.A.; Gühr, M. Understanding the modulation mechanism in resonance-enhanced multiphoton probing of molecular dynamics. Phys. Rev. A 2015, 91. [Google Scholar] [CrossRef]
- Wolf, T.J.A.; Parrish, R.M.; Myhre, R.H.; Martínez, T.J.; Koch, H.; Gühr, M. Observation of Ultrafast Intersystem Crossing in Thymine by Extreme Ultraviolet Time-Resolved Photoelectron Spectroscopy. J. Phys. Chem. A 2019, 123, 6897–6903. [Google Scholar] [CrossRef] [PubMed]
- Werme, L.O.; Bergmark, T.; Siegbahn, K. The High Resolution L2,3 MM and M4,5 NN Auger Spectra from Krypton and M4,5 NN and N4,5 OO Auger Spectra from Xenon. Phys. Scr. 1972, 6, 141–150. [Google Scholar] [CrossRef]
- Braune, M.; Buck, J.; Kuhlmann, M.; Grunewald, S.; Düsterer, S.; Viefhaus, J.; Tiedtke, K. Non-invasive online wavelength measurements at FLASH2 and present benchmark. J. Synchrotron Rad. 2018, 25, 3–15. [Google Scholar] [CrossRef]
- Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules 2017, 22, 998. [Google Scholar] [CrossRef]
- Giuliano, B.M.; Feyer, V.; Prince, K.C.; Coreno, M.; Evangelisti, L.; Melandri, S.; Caminati, W. Tautomerism in 4-Hydroxypyrimidine, S-Methyl-2-thiouracil, and 2-Thiouracil. J. Phys. Chem. A 2010, 114, 12725–12730. [Google Scholar] [CrossRef]
- Föhlisch, A.; Feulner, P.; Hennies, F.; Fink, A.; Menzel, D.; Sanchez-Portal, D.; Echenique, P.M.; Wurth, W. Direct observation of electron dynamics in the attosecond domain. Nature 2005, 436, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.S.; Rumble, C.A.; Vauthey, E. Data analysis in transient electronic spectroscopy—An experimentalist’s view. Int. Rev. Phys. Chem. 2020, 39, 135–216. [Google Scholar] [CrossRef]
- Savelyev, E.; Boll, R.; Bomme, C.; Schirmel, N.; Redlin, H.; Erk, B.; Düsterer, S.; Müller, E.; Höppner, H.; Toleikis, S.; et al. Jitter-correction for IR/UV-XUV pump-probe experiments at the FLASH free-electron laser. New J. Phys. 2017, 19, 043009. [Google Scholar] [CrossRef]
- Dziarzhytski, S.; Biednov, M.; Dicke, B.; Wang, A.; Miedema, P.S.; Engel, R.Y.; Schunck, J.O.; Redlin, H.; Weigelt, H.; Siewert, F.; et al. The TRIXS end-station for femtosecond time-resolved resonant inelastic x-ray scattering experiments at the soft x-ray free-electron laser FLASH. Struct. Dyn. 2020, 7, 054301. [Google Scholar] [CrossRef] [PubMed]
- Trebino, R. Frequency-Resolved Optical Gating: The Measurement of Ultrashort Laser Pulses; Springer Science & Business Media: New York, NY, USA, 2000; ISBN 978-1-4613-5432-1. [Google Scholar]
- Schulz, S.; Grguraš, I.; Behrens, C.; Bromberger, H.; Costello, J.T.; Czwalinna, M.K.; Felber, M.; Hoffmann, M.C.; Ilchen, M.; Liu, H.Y.; et al. Femtosecond all-optical synchronization of an X-ray free-electron laser. Nat. Commun. 2015, 6, 5938. [Google Scholar] [CrossRef]
- Schulz, S.; Czwalinna, M.K.; Felber, M.; Predki, P.; Schefer, S.; Schlarb, H.; Wegner, U. Femtosecond-precision synchronization of the pump-probe optical laser for user experiments at flash. In Proceedings of the Advances in X-ray Free-Electron Lasers ii: Instrumentation, Prague, Czech Republic, 17–19 April 2013; Tschentscher, T., Tiedtke, K., Eds.; Spie-int Soc Optical Engineering: Bellingham, WA, USA, 2013; Volume 8778. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metje, J.; Lever, F.; Mayer, D.; Squibb, R.J.; Robinson, M.S.; Niebuhr, M.; Feifel, R.; Düsterer, S.; Gühr, M. URSA-PQ: A Mobile and Flexible Pump-Probe Instrument for Gas Phase Samples at the FLASH Free Electron Laser. Appl. Sci. 2020, 10, 7882. https://doi.org/10.3390/app10217882
Metje J, Lever F, Mayer D, Squibb RJ, Robinson MS, Niebuhr M, Feifel R, Düsterer S, Gühr M. URSA-PQ: A Mobile and Flexible Pump-Probe Instrument for Gas Phase Samples at the FLASH Free Electron Laser. Applied Sciences. 2020; 10(21):7882. https://doi.org/10.3390/app10217882
Chicago/Turabian StyleMetje, Jan, Fabiano Lever, Dennis Mayer, Richard James Squibb, Matthew S. Robinson, Mario Niebuhr, Raimund Feifel, Stefan Düsterer, and Markus Gühr. 2020. "URSA-PQ: A Mobile and Flexible Pump-Probe Instrument for Gas Phase Samples at the FLASH Free Electron Laser" Applied Sciences 10, no. 21: 7882. https://doi.org/10.3390/app10217882
APA StyleMetje, J., Lever, F., Mayer, D., Squibb, R. J., Robinson, M. S., Niebuhr, M., Feifel, R., Düsterer, S., & Gühr, M. (2020). URSA-PQ: A Mobile and Flexible Pump-Probe Instrument for Gas Phase Samples at the FLASH Free Electron Laser. Applied Sciences, 10(21), 7882. https://doi.org/10.3390/app10217882