Intensive Care in Traumatic Brain Injury Including Multi-Modal Monitoring and Neuroprotection


Abstract
:1. Background
2. Pathological and Pathophysiological Features
3. Traumatic Brain Injury Is Not a Single Entity
4. Multimodal Monitoring and Treatment of Patients with Moderate to Severe TBI
4.1. Neurological Clinical Examination and Early Imaging
4.2. Intracranial Pressure and Cerebral Perfusion Pressure
4.2.1. Intracranial Pressure
4.2.2. Intracranial Pressure Guided “Traditional” Approach
4.2.3. Cerebral Perfusion Pressure Guided Therapy
4.2.4. Rationale for Cerebral Perfusion Pressure Directed Therapy in Traumatic Brain Injuries
- Prevention of ICP rises and maintenance of CPP by basic measures:
- Analgosedation;
- Normocapnic mechanical ventilation;
- Mean systolic arterial pressure of ≥100 mmHg;
- Maintenance of a cerebral perfusion pressure of 60–70 mmHg if autoregulation is preserved;
- Maintenance of a normal body temperature;
- Normoxemia;
- Enteral feeding,
- If ICP increases, active interventions might be warranted:
- Deepening analgosedation;
- Detection and treatment or exclusion of surgically accessible space occupying lesions with imaging;
- Drainage of cerebrospinal fluid (insertion of a ventricular catheter if ventricles are not already fully compressed; risk of infection);
- Increase of mean arterial pressure if autoregulation is preserved in order to reduce cerebral blood volume without compromising cerebral blood flow (risk of cardiovascular side effects, risk of promoting edema formation);
- Use of hyperosmolar solutions (NaCl 3–7.5% (HTS; hypertonic saline), Na–lactate 1100 mosm/L, Mannitol 20%;Hyperosmolar agents are effective in reducing ICP. What remains unanswered is whether these agents contribute toward better neurological outcomes; tiered, algorithmic approaches employed in many trials make it difficult to determine which therapy is beneficial or potentially harmful. Mannitol seems to be less effective than NaCl 7.5% and hypertonic Na–lactate in reducing brain swelling after head injury. Moreover, there is evidence that excessive administration of mannitol may be harmful, because mannitol might pass from the bloodstream into the brain, where it worsens brain edema and increases ICP (rebound edema). In contrast to NaCl 7.5%, mannitol does not improve tissue oxygenation [57]. While both osmolar agents increase cerebral blood flow, the magnitude of augmentation seems to be greater in HTS treatment and HTS seems to reduce the accumulation of extracellular excitatory amino acid (glutamate), thus preventing glutamine toxicity and neuronal damage [58]. However, there is no difference in neurologic outcome between the treatments at 6 months using the Glasgow outcome score [59]. In general, evidence for improved neurological outcome for all these interventions is still very limited, and they bear a risk of fluid overload including cardiovascular events and risks of osmotic diuresis, including dehydration [60].
- Aggressive treatment of refractory elevated intracranial pressure:
- Metabolic suppression (e.g., burst suppression targeted EEG-guided barbiturate coma). Lowers oxygen consumption of the brain by approximately 50% and thus decreases cerebral blood flow and cerebral blood volume, especially if autoregulation is preserved. No proof of improved outcome; risk of cardiovascular depression, risk of infections);
- Hypothermia (e.g., 34 °C; lowers oxygen consumption of the brain and thus cerebral blood flow and cerebral blood volume). Evidence comes mainly from animal experiments; there is no solid beneficial evidence in humans, but an elevated risk for cardiovascular compromise and infections. In a recent multicenter study, it could be shown that in patients with an intracranial pressure of more than 20 mmHg, therapeutic hypothermia plus standard of care to reduce intracranial pressure did not result in better outcomes than standard care alone [61]. Therefore, therapeutic hypothermia is not recommended by the newest guidelines [2];
- Decompressive craniectomy (DC) for selected cases only as DC is a last-resort treatment for severe refractory ICP, reducing ICP and mortality while increasing incidence of unfavorable outcome at six months [62];
- Hypnocapnic ventilation (rescue therapy only; high risk of cerebral ischemia).
4.2.5. Lund Concept: Background and Clinical Application
- Reduction of stress and brain metabolism by analgosedation with low-dose thiopental (0.5–3 mg/kg/h), use of beta-1-antagonist metoprolol, and use of alpha-2-agonist clonidine;
- Reduction of hydrostatic capillary pressure with metoprolol and clonidine;
- Maintenance of colloid-oncotic pressure, control of fluid balance with blood/albumin transfusions and use of furosemide.
- ICP <20–22 mmHg;
- CPP 50–70 mmHg.
4.3. Advanced Bedside Physiological Monitoring for Tailored Management of Cerebral Perfusion Pressure
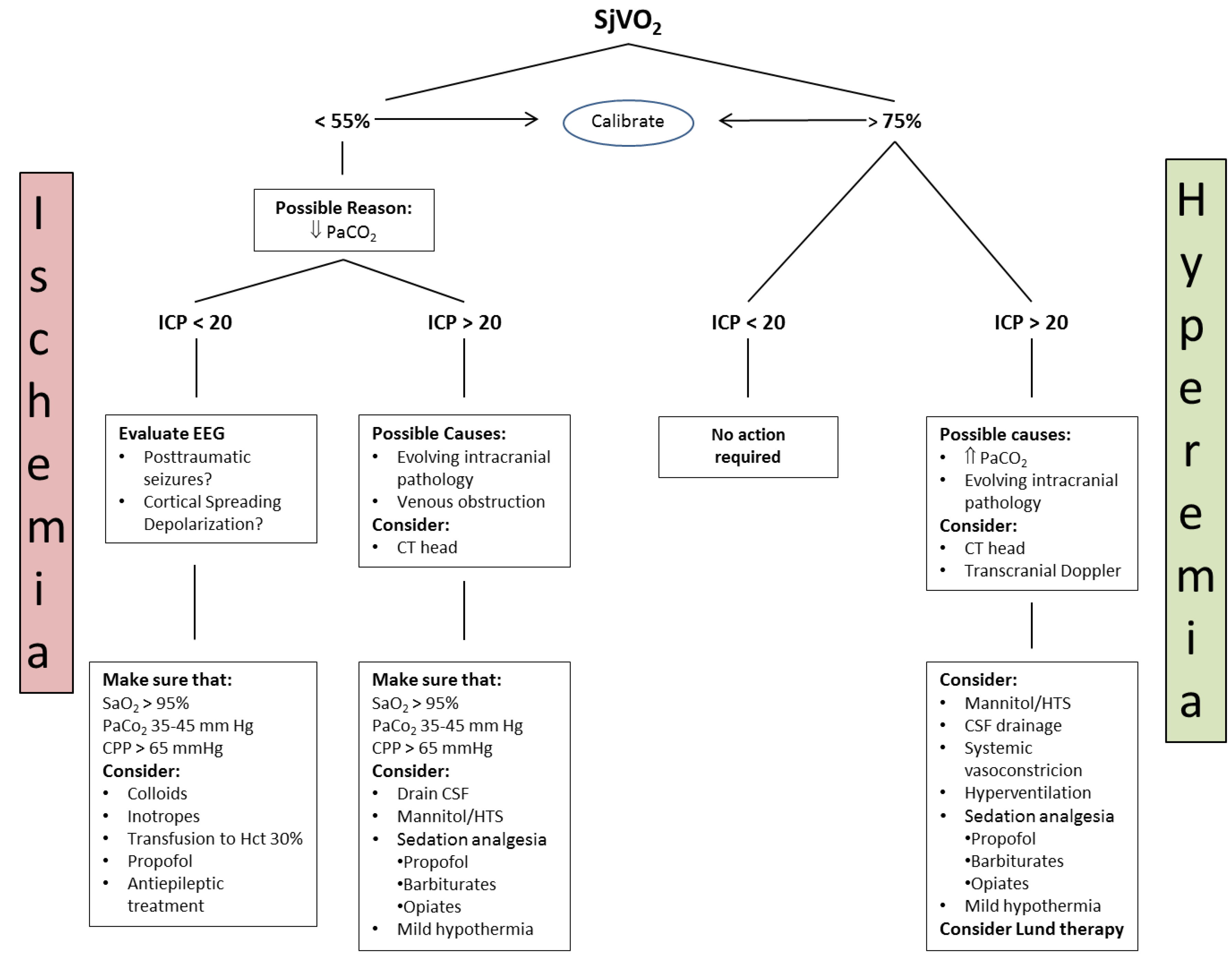
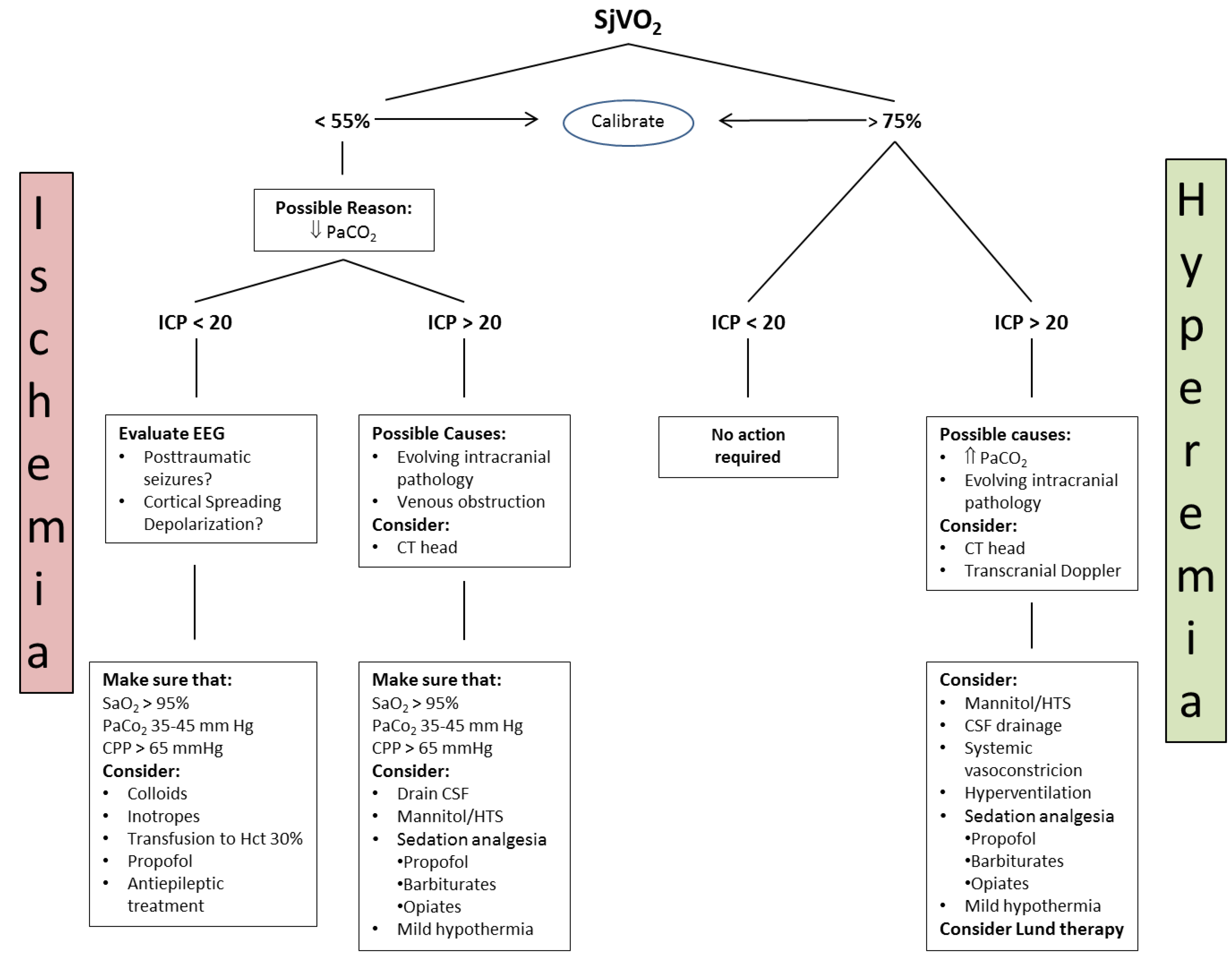
4.3.1. Jugular Bulb Oximetry, Arteriovenous Difference in Lactate
4.3.2. Brain Tissue Partial Tension of Oxygen (PbtO2)
4.3.3. Cerebral Microdialysis
4.3.4. Clinical Use of Cerebral Microdialysis
4.3.5. Cerebral Microdialysis and Seizures, Cortical Spreading Depolarization
4.4. Assessment of Autoregulation
4.5. Simultaneous Multimodal Monitoring for Individualised Management
5. Specific Considerations in Traumatic Brain Injury in the Elderly
6. Conclusions
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention. TBI Data and Statistics. 22 January 2016. Available online: http://www.cdc.gov/traumaticbraininjury/data/ (accessed on 22 February 2019).
- Maas, A.I.R.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratten, S.L.; Chesnut, R.; Harris, O.A.; Kisson, N.; et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, M.A.; Eisenberg, H.M.; Jane, J.A.; Trauma Coma Data Bank Research Group. The Traumatic Coma Data Bank: Design, methods, and baseline caracteristics. J. Neurosurg. 1991, 75, 8–13. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Jackson, T.C.; Ferguson, N.M.; Carlson, S.W.; Simon, D.W.; Brockman, E.C.; Bayir, H.; Poloyac, S.M.; Wagner, A.K.; Kline, A.E.; et al. Emerging therapies in traumatic brain injury. Semin. Neurol. 2015, 35, 83–100. [Google Scholar] [PubMed]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology associated with traumatic brain injury: Current treatments and potential novel therapeutics. Cell. Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Hinzman, J.M.; Wilson, J.A.; Mazzeo, A.T.; Bullok, M.R.; Hartings, J.A. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J. Neurotrauma 2016, 33, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Monro, A. Observations on the Structure and Function of the Nervous System; Creech & Johnson: Edinburgh, UK, 1823; p. 5. [Google Scholar]
- Stocchetti, N.; Maas, A.I. Traumatic intracranial hypertension. N. Engl. J. Med. 2014, 370, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.J.; Sun, M.; Agoston, D.V.; Shultz, S.R. The effect of concomitant peripheral injury on traumatic brain injury pathobiology and outcome. J Neuroinflamm. 2016, 13, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Shotaro Michinaga, S.; Koyama, Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Maas, A.; Lecky, F.; Manley, G.; Stocchetti, N.; Murray, G. The Glasgow Coma Scale at 40 years: Standing the test of time. Lancet Neurol. 2014, 13, 844–854. [Google Scholar] [CrossRef]
- Kurland, D.; Hong, C.; Aarabi, B.; Gerzanich, V. Hemorrhagic Progression of a Contusion after Traumatic Brain Injury: A Review. J. Neurotrauma 2012, 29, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Millar, D.; Jurkovich, G.J. Factor VIIa administration in traumatic brain injury: An AAST-MITC propensity score analysis. Trauma Surg. Acute Care Open 2018, 3, e000134. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Neurological Disorders and Stroke (NINDS) Common Data Elements. Traumatic Brain Injury. March 2017. Available online: https://commondataelements.ninds.nih.gov/TBI.aspx#tab=Data_ Standards (accessed on 22 February 2019).
- Magnoni, S.; Mac Donald, C.L.; Esparza, T.J.; Conte, V.; Sorrell, J.; Macri, M.; Bertani, G.; Biffi, R.; Costa, A.; Sammons, B. Quantitative assessments of traumatic axonal injury in human brain: Concordance of microdialysis and advanced MRI. Brain 2015, 138, 2263–2277. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.; Galarza, M.; Villanueva, P.A. Diffuse axonal injury (DAI) is not associated with elevated intracranial pressure (ICP). Acta Neurochir. 1998, 140, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Miley, J.T.; Rodriguez, G.J.; Qureshi, A.I. Traumatic intracranial aneurysm formation following closed head injury. J. Vasc. Interv. Neurol. 2008, 1, 79–82. [Google Scholar] [PubMed]
- Maas, A.I.R.; Steyerberg, E.W.; Butcher, R.; Damers, J.; Marmarou, L.A.; Mushkudiani, G.; McHugh, G.; Murray, D. Prognostic value of computerized tomography scan characteristics in traumatic brain injury: Results from the IMPACT study. J. Neurotrauma 2007, 24, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; McCormick, W.C.; Kagan, S.H. Traumatic brain injury in older adults: Epidemiology, outcomes, and future implications. J. Am. Geriatr. Soc. 2006, 54, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Paternò, R.; Citerio, G.; Beretta, L.; Colombo, A. Traumatic brain injury in an aging population. J. Neurotrauma 2012, 29, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.A.; Close, J.C. Traumatic brain injury in older adults: Characteristics, causes and consequences. Injury 2012, 43, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Czosnyka, M.; Balestreri, M.; Steiner, L.; Smielewski, P.; Hutchinson, P.J.; Matta, B.; Pickard, J.D. Age, intracranial pressure, autoregulation, and outcome after brain trauma. J. Neurosurg. 2005, 102, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness: A practical scale. Lancet 1974, 304, 81–84. [Google Scholar] [CrossRef]
- Skoglund, K.; Enblad, P.; Hillered, L.; Marklund, N. The neurological wake-up test increases stress hormone levels in patients with severe traumatic brain injury. Crit. Care Med. 2012, 40, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Marklund, N. The Neurological Wake-up Test-A Role in Neurocritical Care Monitoring of Traumatic Brain Injury Patients? Front. Neurol. 2017, 8, 540. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A.; Lu, J.; Butcher, I.; McHugh, G.S.; Murray, G.D.; Steyerberg, E.W.; Mushkudiani, N.A.; Choi, S.; Maas, A.I. Prognostic value of the Glasgow Coma Scale and pupil reactivity in traumatic brain injury assessed pre-hospital and on enrollment: An IMPACT analysis. J. Neurotrauma 2007, 24, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.D.; Behrends, M. Portable infrared pupillometry: A review. Anesth. Analg. 2015, 120, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Juul, N.; Morris, G.F.; Marshall, S.B.; Marshall, L.F. Intracranial hypertension and cerebral perfusion pressure: Influence on neurological deterioration and outcome in severe head injury. The Executive Committee of the International Selfotel Trial. J. Neurosurg. 2000, 92, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.V.; Zada, G.; Salim, A.; Inaba, K.; Kasotakis, G.; Hadjizacharia, P.; Demetriades, D.; Rhee, F. Indications for routine repeat head computed tomography (CT) stratified by severity of traumatic brain injury. J. Trauma 2007, 62, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, E.; Diedler, J.; Kasprowicz, M.; Budohoski, K.P.; Haubrich, C.; Smielewski, P.; Outtrim, J.G.; Manktelow, A.; Hutchinson, P.J.; Pickard, J.D.; Menon, D.K.; Czosnyka, M. Critical thresholds for cerebrovascular reactivity after traumatic brain injury. Neurocrit. Care 2012, 16, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Chesnut, R.M.; Temkin, N.; Carney, N.; Dikmen, S.; Rondina, C.; Videtta, W.; Petroni, G.; Lujan, S.; Pridgeon, J.; Barber, J.; Machamer, J.; Chaddock, K. A trial of intracranial-pressure monitoring in traumatic brain injury. N. Engl. J. Med. 2012, 367, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Chesnut, R.M.; Clifton, G.; Ghajar, J.; Marion, D.W.; Narayan, R.K.; Newell, D.W.; Pitts, L.H.; Rosner, M.J.; Wilberger, J.W. Brain Trauma Foundation, American Association of Neurological Surgeons, and Joint Section on Neurotrauma and Critical Care. Guidelines for the management of severe head injury. J. Neurotrauma 1996, 13, 641–734. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Georgoff, P.; Meghan, S.; Mirza, K.L.; El Falaky, O.M. Relationship of aggressive monitoring and treatment to improved outcomes in severe traumatic brain injury. J. Neurosurg. 2010, 112, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.C.; Bouamra, O.; Woodford, M.; King, A.T.; Yates, D.W.; Lecky, F.E. Trends in head injury outcome from 1989 to 2003 and the effect of neurosurgical care: An observational study. Lancet 2005, 366, 1538–1544. [Google Scholar] [CrossRef]
- Marmarou, A.; Saad, A.; Aygok, G.; Rigsbee, M. Contribution of raised ICP and hypotension to CPP reduction in severe brain injury: Correlation to outcome. Acta Neurochir. Suppl. 2005, 95, 277–280. [Google Scholar] [PubMed]
- Narayan, R.K.; Kishore, P.R.; Becker, D.P.; Ward, J.D.; Enas, G.G.; Greenberg, R.P.; Domingues Da Silva, A.; Lipper, M.H.; Choi, S.C.; Mayhall, C.G.; Lutz, H.A.; Young, H.F. Intracranial pressure: To monitor or not to monitor? A review of our experience with severe head injury. J. Neurosurg. 1982, 56, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Piper, I.R.; Dearden, N.M. Management of intracranial hypertension in head injury: Matching treatment with cause. Acta Neurochir. Suppl. 1993, 57, 152–159. [Google Scholar] [PubMed]
- Young, J.S.; Blow, O.; Turrentine, F.; Claridge, J.A.; Schulman, A. Is there an upper limit of intracranial pressure in patients with severe head injury if cerebral perfusion pressure is maintained? Neurosurg. Focus 2003, 15, E2. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.C.; Radinsky, C.R.; Furlan, A.J.; Chyatte, D.; Qu, Y.; Easley, K.A.; Perez-Trepichio, A.D. Variability in the magnitude of the cerebral blood flow response and the shape of the cerebral blood flow-pressure autoregulation curve during hypotension in normal rats [corrected]. Anesthesiology 2002, 97, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Czosnyka, M.; Smielewski, P.; Piechnik, S.; Steiner, L.A.; Pickard, J.D. Cerebral autoregulation following head injury. J. Neurosurg. 2001, 95, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.J.; Rosner, S.D.; Johnson, A.H. Cerebral perfusion pressure: Management protocol and clinical results. J. Neurosurg. 1995, 83, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Asgeirsson, B.; Grande, P.O.; Nordstrom, C.H. A new therapy of post-trauma brain oedema based on haemodynamic principles for brain volume regulation. Intensive Care Med. 1994, 20, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.J.; Daughton, S. Cerebral perfusion pressure management in head injury. J. Trauma 1990, 30, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Contant, C.F.; Valadka, A.B.; Gopinath, S.P.; Hanay, H.J.; Robertson, C.S. Adult respiratory distress syndrome: A complication of induced hypertension after severe head injury. J. Neurosurg. 2001, 95, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.L.; Miller, E.R.; Choi, S.C.; Levin, H.S. Fluid thresholds and outcome from severe brain injury. Crit. Care Med. 2002, 30, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.J.; Muizelaar, J.P. Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation. J. Neurosurg. 1990, 73, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.J.; Muizelaar, J.P.; Bandoh, K.; Marmarou, A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: Relationship with cerebral blood flow. J. Neurosurg. 1992, 77, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Kiening, K.L.; Hartl, R.; Unterberg, A.W.; Schneider, G.H.; Bardt, T.; Lanksch, W.R. Brain tissue pO2-monitoring in comatose patients: Implications for therapy. Neurol. Res. 1997, 19, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.S.; Valadka, A.B.; Hannay, H.J.; Contant, C.F.; Gopinath, S.P.; Cormio, M.; Uzura, M.; Grossman, R.G. Prevention of secondary ischemic insults after severe head injury. Crit. Care Med. 1999, 27, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, C.H. Assessment of critical thresholds for cerebral perfusion pressure by performing bedside monitoring of cerebral energy metabolism. Neurosurg. Focus 2003, 15, E5. [Google Scholar] [CrossRef] [PubMed]
- Carrera, E.; Kim, D.J.; Castellani, G.; Zweifel, C.; Czosnyka, Z.; Kasparowicz, M.; Smielewski, P.; Pickard, J.D.; Czosnyka, M. What shapes pulse amplitude of intracranial pressure? J. Neurotrauma 2010, 27, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Neff, T.A.; Doelberg, M.; Jungheinrich, C.; Sauerland, A.; Spahn, D.R.; Stocker, R. Repetitive large-dose infusion of the novel hydroxyethyl starch 130/0.4 in patients with severe head injury. Anesth. Analg. 2003, 96, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- The SAFE Study Investigators. Saline or Albumin for Fluid Resuscitation in Patients with Traumatic Brain Injury. N. Engl. J. Med. 2007, 357, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Unterberg, A.W.; Kiening, K.L.; Härtl, R.; Bardt, T.; Sarrafzadeh, A.S.; Lanksch, W.R. Multimodal monitoring in patients with head injury: Evaluation of the effects of treatment on cerebral oxygenation. J. Trauma 1997, 42, 32–37. [Google Scholar] [CrossRef]
- Cottenceau, V.; Masson, F.; Mahamid, E.; Petit, L.; Shik, V.; Sztark, F.; Zaaroor, M.; Soustiel, J.F. Comparison of effects of equiosmolar doses of mannitol and hypertonic saline on cerebral blood flow and metabolism in traumatic brain injury. J. Neurotrauma 2011, 28, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Boone, M.D.; Oren-Grinberg, A.; Robinson, T.M.; Chen, C.C.; Kasper, E.M. Mannitol or hypertonic saline in the setting of traumatic brain injury: What have we learned? Surg. Neurol. Int. 2015, 6, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Stover, J.F.; Steiger, P.; Stocker, R. Treating Intracranial Hypertension in Patients with Severe Traumatic Brain Injury during neurointensive care. Eur. J. Trauma 2005, 31, 308–330. [Google Scholar] [CrossRef]
- Andrews, P.J.D.; Sinclair, L.; Rodriguez, A.; Harris, B.A.; Battison, C.G.; Rhodes, J.K.J.; Murray, G.D. Hypothermia for Intracranial Hypertension after Traumatic Brain Injury. N. Engl. J. Med. 2015, 373, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.; Dutton, R.; Aarabi, B.; Scalea, T. Decompressive Laparotomy to Treat Intractable Intracranial Hypertension after Traumatic Brain Injury. J. Trauma 2004, 57, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, C.H. Physiological and biochemical principles underlying volume-targeted therapy—The “Lund concept”. Neurocrit. Care 2005, 2, 83–95. [Google Scholar] [CrossRef]
- Eker, C.; Asgeirsson, B.; Grände, P.O. Improved outcome after severe head injury with a new therapy based on principles for brain volume regulation and preserved microcirculation. Crit. Care Med. 1998, 26, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Grande, P.O.; Asgeirsson, B.; Nordstorm, C.H. Physiologic principles for volume regulation of a tissue enclosed in a rigid shell with application to the injured brain. J. Trauma 1997, 42, 23–31. [Google Scholar] [CrossRef]
- Liu, C.W.; Zeng, Y.K.; Lu, J.; Yu, W.H.; Wang, B.; Hu, W.; Zhu, K.Y.; Zhu, Y.; Hu, W.H.; et al. Application of the Lund concept in treating brain edema after severe head injury. Zhongguo Wei Zong Bing Ji Jiu Yi Xue 2010, 22, 610–613. [Google Scholar]
- Dizdarevic, K.; Hamdan, A.; Omerhodzic, I.; Kominlilja-Smajic, E. Modified Lund concept versus cerebral perfusion pressure-targeted therapy: A randomised controlled study in patients with secondary brain ischemia. Clin. Neurol. Neurosurg. 2012, 114, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Muzevic, D.; Splavski, B. The Lund concept for severe traumatic brain injury (Review). Cochrane Database Syst. Rev. 2013, CD010193. [Google Scholar] [CrossRef]
- Roberts, I.; Yates, D.; Sandercock, P.; Farrell, B.; Wasserberg, J.; Lomas, G.; Cottingham, R.; Svoboda, P.; Brayley, N.; Mazairac, G.; et al. Effect of intravenous corticosteroids on death within 14 days in 10 008 adults with clinically significant head injury: Randomised placebo-controlled trial. Lancet 2004, 364, 1321–1328. [Google Scholar] [PubMed]
- Bratton, S.L.; Chestnut, R.M.; Ghajar, J.; McConnell, F.F.H.; Harris, O.A.; Hartl, R.; Manley, G.T.; Nemecek, A.; Newell, D.W.; Rosenthal, G.; et al. Guidelines for the management of severe traumatic brain injury. IX. Cerebral perfusion thresholds. J. Neurotrauma 2007, 24 (Suppl. 1), S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, P.; Menon, D.K.; Citerio, G.; Vespa, P.; Bader, M.K.; Brophy, G.M.; Diringer, M.N.; Stocchetti, N.; Videtta, W.; Armonda, R.; et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care: A statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Intensive Care Med. 2014, 40, 1189–1209. [Google Scholar] [PubMed]
- Brandi, G.; Bechir, M.; Sailer, S.; Haberthür, C.; Stocker, R.; Stover, J.F. Transcranial color-coded duplex sonography allows to assess cerebral perfusion pressure noninvasively following severe traumatic brain injury. Acta Neurochir. 2010, 152, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Wijayatilake, D.S.; Talati, C.; Panchatsharam, S. The monitoring and management of severe traumatic brain injury in the United Kingdom: Is there a consensus? A national survey. J. Neurosurg. Anesthesiol. 2015, 27, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.E.; Charbel, F.T.; Edelman, G.; Ausman, J.I. Brain tissue oxygen pressure, carbon dioxide pressure and pH during ischemia. Neurol. Res. 1996, 18, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J. The first decade of continuous monitoring of jugular bulb oxyhemoglobin saturation: Management strategies and clinical outcome. Crit. Care Med. 1998, 26, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.M.; Chan, M.S.; Poon, W.S. Cerebral venous oxygen saturation monitoring: Is dominant jugular bulb cannulation good enough? Br. J. Neurosurg. 1996, 10, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.S.; Narayan, R.K.; Gokaslan, Z.L.; Pahwa, R.; Grossman, R.G.; Caram, P. Jr.; Allen, E. Cerebral arteriovenous oxygen difference as an estimate of cerebral blood flow in comatose patients. J. Neurosurg. 1989, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Schurer, L. Correlation of jugular venous oxygen saturation to spontaneous fluctuations of cerebral perfusion pressure in patients with severe head injury. Neurol. Res. 1995, 17, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Chieregato, A.; Marchi, M.; Zoppellari, R.; Fabbri, E.; Cianchi, G.; Forini, E.; Targa, L. Detection of early ischemia in severe head injury by means of arteriovenous lactate differences and jugular bulb oxygen saturation. Relationship with CPP, severity indexes and outcome. Preliminary analysis. Acta Neurochir. 2002, 81, 289–293. [Google Scholar]
- Gupta, A.K.; Zygun, D.A.; Johnston, A.J.; Steiner, L.A.; Al-Rawi, P.G.; Chatfield, D.; Shepherd, E.; Kirkpatrick, P.J.; Hutchinson, P.J.; Menon, D.K. Extracellular Brain pH and Outcome following Severe Traumatic Brain Injury. J. Neurotrauma 2004, 21, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Coles, J.P.; Fryer, T.D.; Smielewski, P.; Chatfield, D.A.; Steiner, L.A.; Johnston, A.J.; Downey, S.P.; Williams, G.B.; Aigbirhio, F.; Hutchinson, P.J.; et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J. Cereb. Blood Flow Metab. 2004, 24, 202–211. [Google Scholar] [CrossRef] [PubMed]
- White, H.; Baker, A. Continuous jugular venous oximetry in the neurointensive care unit—A brief review. Can. J. Anaesth. 2002, 49, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.A.; Barletta, J.F.; Figueroa, B.E.; Bonnell, B.W.; Vanderkolk, W.E.; McAllen, K.J.; Ott, M.M. Cerebral perfusion pressure and intracranial pressure are not surrogates for brain tissue oxygenation in traumatic brain injury. Clin. Neurophysiol. 2012, 123, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- van Santbrink, H.; vd Brink, W.A.; Steyerberg, E.W.; Carmona Suazo, J.A.; Avezaat, C.J.; Maas, A.I. Brain tissue oxygen response in severe traumatic brain injury. Acta Neurochir. (Wien) 2003, 145, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P. What is the optimal threshold for cerebral perfusion pressure following traumatic brain injury? Neurosurg. Focus 2003, 15, E4. [Google Scholar] [CrossRef] [PubMed]
- Jodicke, A.; Hubner, F.; Boker, D.K. Monitoring of brain tissue oxygenation during aneurysm surgery: Prediction of procedure-related ischemic events. J. Neurosurg. 2003, 98, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Castilla, L.; Lara, L.R.; Gopinath, S.; Swank, P.R.; Valadka, A.; Robertson, C. Cerebral hemodynamic effects of acute hyperoxia and hyperventilation after severe traumatic brain injury. J. Neurotrauma 2010, 27, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, W.A.; van Santbrink, H.; Avezaat, C.J.J.; Hogesteeger, C.; Jansen, W.; Kloos, L.M.; Vermeulen, J.; Mass, A.I. Monitoring brain oxygen tension in severe head injury: The Rotterdam experience. Acta Neurochir. Suppl. 1998, 71, 190–194. [Google Scholar] [PubMed]
- Rosenthal, G.; Hemphill, J.C.I.I.I.; Sorani, M.; Martin, C.; Morabito, D.; Obrist, W.D.; Manley, G.T. Brain tissue oxygen tension is more indicative of oxygen diffusion than oxygen delivery and metabolism in patients with traumatic brain injury. Crit. Care Med. 2008, 36, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.K.; Coles, J.P.; Gupta, A.K.; Fryer, T.D.; Smielewski, P.; Chatfield, D.A.; Aigbirhio, F.; Skepper, J.N.; Minhas, P.S.; Hutchinson, P.J.; Carpenter, T.A.; et al. Diffusion limited oxygen delivery following head injury. Crit. Care Med. 2004, 32, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Meixensberger, J.; Jaeger, M.; Väth, A.; Dings, J.; Kunze, E.; Roosen, K. Brain tissue oxygen guided treatment supplementing ICP/CPP therapy after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2003, 74, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Spiotta, A.M.; Stiefel, M.F.; Gracias, V.H.; Garuffe, A.M.; Kofke, W.A.; Maloney-Wilensky, E.; Troxel, A.B.; Levine, J.M.; Le Roux, P.D. Brain tissue oxygen-directed management and outcome in patients with severe traumatic brain injury. J. Neurosurg. 2010, 113, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Soehle, M.; Jaeger, M.; Meixensberger, J. Online assessment of brain tissue oxygen autoregulation in traumatic brain injury and subarachnoid hemorrhage. Neurol. Res. 2003, 25, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, M.L.; Durham, C.A.; McNally, M.M.; Agle, S.C.; Parker, F.M.; Stoner, M.C. Correlation of cerebral oximetry with internal carotid artery stump pressures in carotid endarterectomy. Vasc. Endovasc. Surg. 2010, 44, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Sahuquillo, J.; Amoros, S.; Santos, A.; Poca, M.A.; Panzardo, H.; Domínguez, L.; Pedraza, S. Does an increase in cerebral perfusion pressure always mean a better oxygenated brain? A study in head-injured patients. Acta Neurochir. Suppl. 2000, 76, 457–462. [Google Scholar] [PubMed]
- Stiefel, M.F.; Spiotta, A.; Gracias, V.H.; Garuffe, A.M.; Guillamondegui, O.; Maloney-Wilensky, E.; Bloom, S.; Grady, M.S.; LeRoux, P.D. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J. Neurosurg. 2005, 103, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Adamides, A.A.; Cooper, D.J.; Rosenfeldt, F.L.; Bailey, M.J.; Pratt, N.; Tippett, N.; Vallance, S.; Rosenfeld, J.V. Focal cerebral oxygenation and neurological outcome with or without brain tissue oxygen-guided therapy in patients with traumatic brain injury. Acta Neurochir. (Wien) 2009, 151, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Narotam, P.K.; Burjonrappa, S.C.; Raynor, S.C.; Rao, M.; Taylon, C. Cerebral oxygenation in major pediatric trauma: Its relevance to trauma severity and outcome. J. Pediatr. Surg. 2006, 41, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Narotam, P.K.; Morrison, J.F.; Nathoo, N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: Outcome analysis of a brain tissue oxygen-directed therapy. J. Neurosurg. 2009, 111, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Martini, R.P.; Deem, S.; Yanez, N.D.; Chesnut, R.M.; Weiss, N.S.; Daniel, S.; Souter, M.; Treggiari, M.M. Management guided by brain tissue oxygen monitoring and outcome following severe traumatic brain injury. J. Neurosurg. 2009, 111, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Dengl, M.; Meixensberger, J.; Schuhmann, M.U. Effects of cerebrovascular pressure reactivity-guided optimization of cerebral perfusion pressure on brain tissue oxygenation after traumatic brain injury. Crit. Care Med. 2010, 38, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Nangunoori, R.; Maloney-Wilensky, E.; Stiefel, M.; Park, S.; Andrew Kofke, W.; Levine, J.M.; Yang, W.; Le Roux, P.D. Brain tissue oxygen-based therapy and outcome after severe traumatic brain injury: A systematic literature review. Neurocrit. Care 2012, 17, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, I.; Carpenter, K.L.; Nortje, J.; Al-Rawi, P.G.; O'Connell, M.T.; Czosnyka, M.; Smielewski, P.; Pickard, J.D.; Menon, D.K.; Kirkpatrick, P.J.; et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: A microdialysis study of 223 patients. Brain 2011, 134, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Quintard, H.; Patet, C.; Zerlauth, J.B.; Suys, T.; Bouzat, P.; Pellerin, L.; Meuli, R.; Magistretti, P.J.; Oddo, M. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J. Neurotrauma 2015, 33, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zauner, A.; Doppenberg, E.M.; Woodward, J.J.; et al. Continuous monitoring of cerebral substrate delivery and clearance: Initial experience in 24 patients with severe acute brain injuries. Neurosurgery 1997, 41, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Zauner, A.; Doppenberg, E.; Woodward, J.J.; Choi, S.C.; Young, H.F.; Bullock, R. Multiparametric continuous monitoring of brain metabolism and substrate delivery in neurosurgical patients. Neurol. Res. 1997, 19, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.C.; Robertson, C.S. Microdialysis: Is it ready for prime time? Curr. Opin. Crit. Care 2009, 15, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Tisdall, M.M.; Smith, M. Cerebral microdialysis: Research technique or clinical tool. Br. J. Anaesth. 2006, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bor-Seng-Shu, E.; Oliveira, M.L.; Teixeira, M.J. Traumatic brain injury and metabolism. J. Neurosurg. 2010, 112, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Peerdeman, S.M.; Girbes, A.R.J.; Vandertop, W.P. Cerebral microdialysis as a new tool for neurometabolic monitoring. Intensive Care Med. 2000, 26, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Nordström, C.H. Cerebral energy metabolism and microdialysis in neurocritical care. Childs Nerv. Sys. 2010, 26, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Belli, A.; Sen, J.; Petzold, A.; Russo, S.; Kitchen, N.; Smith, M. Metabolic failure precedes intracranial pressure rises in traumatic brain injury: A microdialysis study. Acta Neurochir. (Wien) 2008, 150, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Mellergard, P.; Hasllstrom, A.; Ungerstedt, U.; Nordström, C.H. Intracerebral microdialysis and bedside biochemical analysis in patients with fatal traumatic brain lesions. Acta Anaesthsiol. Scand. 2013, 45, 977–985. [Google Scholar] [CrossRef]
- Timofeev, I.; Czosnyka, M.; Carpenter, K.L.H.; Nortje, J.; Kirkpatrick, P.J.; Al-Rawi, P.G.; Menon, D.K.; Pickard, J.D.; Gupta, A.K.; Hutchinson, P.J. Interaction between brain chemistry and physiology after traumatic brain injury: Impact of autoregulation and microdialysis catheter location. J. Neurotrauma 2011, 28, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.I.; Stiefel, M.F.; Oddo, M.; Milby, A.H.; Maloney-Wilensky, E.; Frangos, S.; Levine, J.M.; Kofke, W.A.; LeRoux, P.D. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery 2011, 69, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Ungerstedt, U.; Nordstrom, C.H. Brain energy metabolism during controlled reduction of cerebral perfusion pressure in severe head injuries. Intensive Care Med. 2001, 27, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.J.; Steiner, L.A.; Chatfield, D.A.; Coles, J.P.; Hutchinson, P.J.; Al-Rawi, P.G.; Menon, D.K.; Gupta, A.K. Effect of cerebral perfusion pressure augmentation with dopamine and norepinephrine on global and focal brain oxygenation after traumatic brain injury. Intensive Care Med. 2004, 30, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Veenith, T.V.; Carter, E.L.; Grossac, J.; Newcombe, V.F.; Outtrim, J.G.; Nallapareddy, S.; Lupson, V.; Correia, M.M.; Mada, M.M.; Williams, G.B.; et al. Use of diffusion tensor imaging to assess the impact of normobaric hyperoxia within at-risk pericontusional tissue after traumatic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.; Boonyaputthiku, R.; MacArthur, D.L.; Miller, C.; Etchepare, M.; Bergsneider, M.; Glenn, T.; Martin, N.; Hovda, D. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. 2006, 34, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Meierhans, R.; Bechir, M.; Ludwig, S.; Sommerfeld, J.; Brandi, G.; Haberthür, C.; Stocker, R.; Stover, J.F. Brain metabolism is significantly impaired at blood glucose below 6 mM and brain glucose below 1 mM in patients with severe traumatic brain injury. Crit. Care 2010, 14, R13. [Google Scholar] [CrossRef] [PubMed]
- Oddo, M.; Schmidt, J.M.; Carrera, E.; Badjatia, N.; Connolly, E.S.; Presciutti, M.; Ostapkovich, N.D.; Levine, J.M.; Le Roux, P.; Mayer, S.A. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: A microdialysis study. Crit. Care 2008, 36, 3233–3238. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.; Prins, M.; Ronne-Engstrom, E.; Caron, M.; Shalmon, E.; Hovda, D.A.; Martin, N.A.; Becker, D.P. Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: A microdialysisstudy. J. Neurosurg. 1998, 89, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.M.; Miller, C.; McArthur, D.; Eliseo, M.; Etchepare, M.; Hirt, D.; Glenn, T.C.; Martin, N.; Hovda, D. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit. Care 2007, 35, 2830–2836. [Google Scholar] [CrossRef]
- Hillerd, L.; Persson, L.; Nilsson, P.; et al. Continuous monitoring of cerebral metabolism in traumatic brain injury: A focus on cerebral microdialysis. Curr. Opin. Crit. Care 2006, 12, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Wartenberg, K.E. Critical care of poor-grade subarachnoid hemorrhage. Curr. Opin. Crit. Care 2011, 17, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bergsneider, M.; Hovda, D.A.; Shalmon, E.; Kelly, D.F.; Vespa, P.M.; Martin, N.A.; Phelps, M.E.; McArthur, D.L.; Caron, M.J.; Kraus, J.F.; et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J. Neurosurg. 1997, 86, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sunami, K.; Nakamura, T.; Kubota, M.; Yoshinori, O.; Hiroki, N.; Akira, Y.; Hiroyasu, M. Spreading depression following experimental head injury in the rat. Neurol. Med. Chir. 1989, 29, 975–980. [Google Scholar] [CrossRef]
- Hopwood, S.E.; Parkin, M.C.; Bezzina, E.L.; Boutelle, M.G.; Strong, A.J. Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis. J. Cereb. Blood Flow Metab. 2005, 25, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.C.; Beltran-Parrazal, L.; Lopez-Valdes, H.E.; Theriot, J.; Toga, A.W.; Charles, A.C. Distinct vascular conduction with cortical spreading depression. J. Neurophysiol. 2007, 97, 4143–4151. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, M.; Fuhr, S.; Bhatia, R.; Boutelle, M.; Hashemi, P.; Strong, A.J.; Lauritzen, M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006, 129, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Steiner, L.A.; Czosnyka, M.; Piechnik, S.K.; Smielewski, P.; Chatfield, D.; Menon, D.K.; Pickard, J.D. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit. Care Med. 2002, 30, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Aries, M.J.; Czosnyka, M.; Budohoski, K.P.; Steiner, L.A.; Lavinio, A.; Kolias, A.G.; Hutchinson, P.J.; Brady, K.M.; Menon, D.K.; Pickard, J.D. Continuous determination of optimal cerebral perfusion pressure in traumatic brain injury. Crit. Care Med. 2012, 40, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.; Bergsneider, M.; Hattori, N.; Wu, H.M.; Huang, S.C.; Martin, N.A.; Glenn, T.C.; McArthur, D.L.; Hovda, D.A. Metabolic crisis without brain ischemia is common after traumatic brain injury: A combined microdialysis and positron emission tomography study. J. Cereb. Blood Flow Metab. 2005, 25, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Magnoni, S.; Ghisoni, L.; Locatelli, M.; Caimi, M.; Colombo, A.; Valeriani, V.; Stocchetti, N. Lack of improvement in cerebral metabolism after hyperoxia in severe head injury: A microdialysis study. J. Neurosurg. 2003, 98, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Vespa, P.M.; McArthur, D.; O’Phelan, K.; Glenn, T.; Etchepare, M.; Kelly, D.; Bergsneider, M.; Martin, N.A.; Hovda, D.A. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: A microdialysis study. J. Cereb. Blood Flow Metab. 2003, 23, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Holbein, M.; Béchir, M.; Ludwig, S.; Sommerfeld, J.; Cottini, S.R.; Keel, M.; Stocker, R.; Stover, J.F.; Sommerfeld, J.; Cottini, S.R.; Keel, M.; Stocker, R.; Stover, J.F. Differential influence of arterial blood glucose on cerebral metabolism following severe traumatic brain injury. Crit. Care 2009, 13, R13. [Google Scholar] [CrossRef] [PubMed]
- Güiza, F.; Depreitere, B.; Piper, I.; Citerio, G.; Chambers, I.; Jones, P.A.; Lo, T.Y.; Enblad, P.; Nillson, P.; Feyen, B.; et al. Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med. 2015, 41, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Aries, M.J.; Wesselink, R.; Elting, J.W.; Donnelly, J.; Czosnyka, M.; Ercole, A.; Maurits, N.M.; Smielewski, P. Enhanced visualization of optimal cerebral perfusion pressure over time to support clinical decision making. Crit. Care Med. 2016, 44, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Dams-O’Connor, K.; Cuthbert, J.P.; Whyte, J.; Corrigan, J.D.; Faul, M.; Harrison-Felix, C. Traumatic brain injury among older adults at level I and II trauma centers. J. Neurotrauma 2013, 30, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, D.M.; McWilliams, N.; Schatz, P.; Hillary, F.G. Epidemiological shifts in elderly traumatic brain injury: 18-year trends in Pennsylvania. J. Neurotrauma 2012, 29, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Hollis, S.; Lecky, F.; Yates, D.W.; Woodford, M. The effect of pre-existing medical conditions and age on mortality after injury. J. Trauma 2006, 61, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Woolcott, J.C.; Richardson, K.J.; Wiens, M.O.; Patel, B.; Marin, J.; Khan, K.M.; Marra, C.A. Meta-analysis of the impact of 9 medication classes of falls in elderly persons. Arch. Intern. Med. 2009, 169, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Gaist, D.; García Rodríguez, L.A.; Hellfritzsch, M.; Poulsen, F.R.; Halle, B.; Hallas, J.; Pottegård, A. Association of antithrombotic drug use with subdural hematoma risk. JAMA 2017, 317, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, A.; Smith, J.E.; Bouamra, O.; Edwards, A.; Yates, D.; Lecky, F. Older patients with traumatic brain injury present with a higher GCS score than younger patients for a given severity of injury. Emerg. Med. J. 2016, 33, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Caterino, J.M.; Raubenolt, A.; Cudnik, M.T. Modification of Glasgow Coma Scale criteria for injured elders. Acad. Emerg. Med. 2011, 18, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Mathias, J.L.; Wheaton, P. Contribution of brain or biological reserve and cognitive or neural reserve to outcome after TBI: A meta-analysis (prior to 2015). Neurosci. Biobehav. Rev. 2015, 55, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Jenks, T.; Bouamra, O.; Edwards, A.; Yates, D.; Wilson, M.H. Increased mortality associated with cerebral contusions following trauma in the elderly: Bad patients or bad management? J. Neurotrauma 2013, 30, 1385–1390. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Intervention | Effect | Pot. Benefit | Pot. Risk |
|---|---|---|---|
| (Deepening of) Analgosedation, Barbiturates | Decrease of brain metabolism and therefore oxygen consumption | Decrease in CBF, CBV and ICP | Adverse effects of long-term sedation, impaired neurological assessment |
| Osmotherapy with Mannitol | Reduction in brain tissue volume, increase in CBF | Decreases ICP, no improvement of tissue oxygenation | Fluid overload, osmotic diuresis leading to hypovolemia, hyperosmolarity, rebound brain edema |
| Osmotherapy with hypertonic saline | Reduction in brain tissue volume, increase in CBF | Decreases ICP, improvement of tissue oxygenation | Fluid overload, osmotic diuresis leading to hypovolemia, hyperosmolarity |
| CSF drainage | Allows for expansion of brain tissue by reducing CSF volume | Lowers ICP | Transcerebral ventricular drainage required, risk of ventriculitis, meningitis |
| Hyperventilation | Cerebral vasoconstriction leading to reduced CBV | Lowers ICP if autoregulation is intact | Reduction in CBF leading to brain tissue ischemia |
| Decompressive craniectomy | Allows for expansion of the edematous brain | Decreases ICP, may improve tissue perfusion | Aggravation of brain edema, unfavorable outcome after 6 months |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stocker, R.A. Intensive Care in Traumatic Brain Injury Including Multi-Modal Monitoring and Neuroprotection. Med. Sci. 2019, 7, 37. https://doi.org/10.3390/medsci7030037
Stocker RA. Intensive Care in Traumatic Brain Injury Including Multi-Modal Monitoring and Neuroprotection. Medical Sciences. 2019; 7(3):37. https://doi.org/10.3390/medsci7030037
Chicago/Turabian StyleStocker, Reto A. 2019. "Intensive Care in Traumatic Brain Injury Including Multi-Modal Monitoring and Neuroprotection" Medical Sciences 7, no. 3: 37. https://doi.org/10.3390/medsci7030037
APA StyleStocker, R. A. (2019). Intensive Care in Traumatic Brain Injury Including Multi-Modal Monitoring and Neuroprotection. Medical Sciences, 7(3), 37. https://doi.org/10.3390/medsci7030037