Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis

{kind=link}
{kind=link}
Abstract
1. Introduction
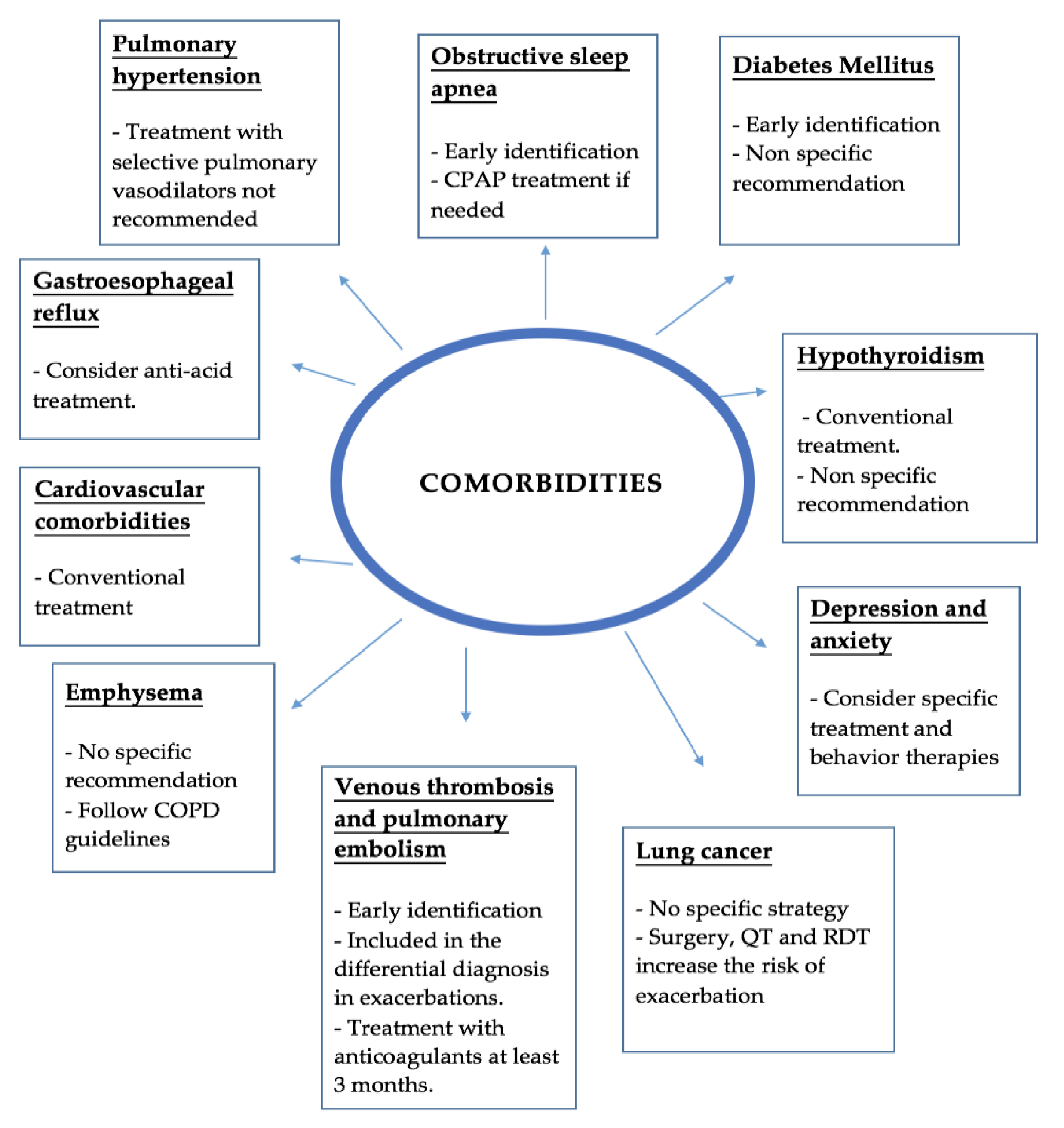
2. Comorbidities
2.1. Pulmonary Hypertension
2.2. Emphysema
2.3. Obstructive Sleep Apnea
2.4. Gastroesophageal Reflux
2.5. Cardiovascular Comorbidities
2.6. Lung Cancer
2.7. Venous Thrombosis and Pulmonary Embolism
2.8. Diabetes Mellitus
2.9. Hypothyroidism
2.10. Anxiety and Depression
3. Complications
Acute Exacerbation
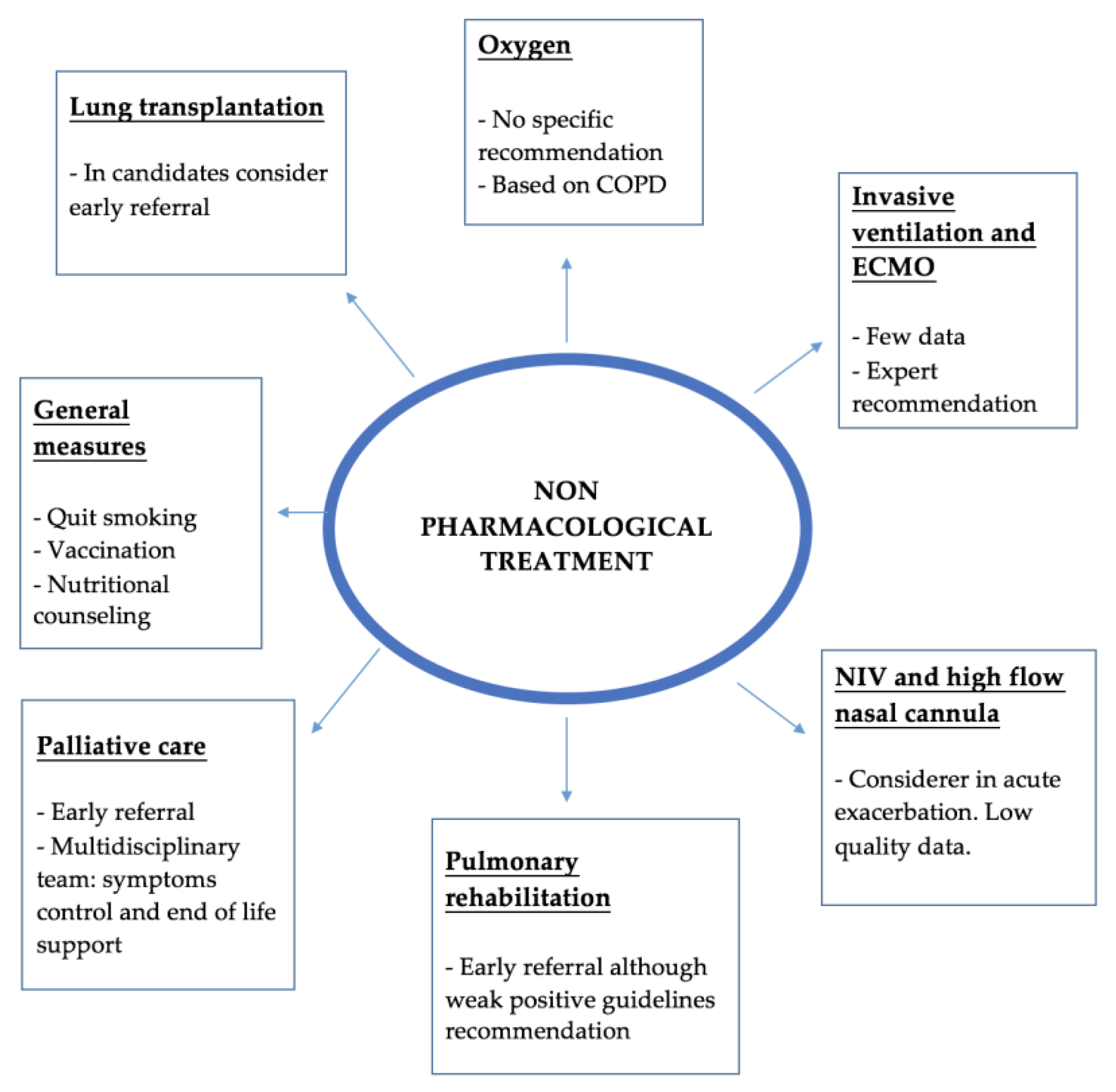
4. Non-Pharmacological Treatment
4.1. General Measures
4.2. Oxygen
4.3. Support Therapies in Acute Life-Threating Hospitalizations
4.4. Lung Transplantation
- Histopathologic or radiographic evidence of usual interstitial pneumonitis or fibrosing non-specific interstitial pneumonitis, regardless of lung function.
- Abnormal lung function with FVC <80% predicted or DLCO <40% predicted.
- Any dyspnea or functional limitation attributable to lung disease.
- Any oxygen requirement, even if only during exertion.
- For inflammatory interstitial lung disease (ILD); failure to improve dyspnea, oxygen requirement, and/or lung function after a clinically indicated trial of medical therapy.
- Decline in FVC >10% during six months of follow-up.
- Decline in DLCO >15% during six months of follow-up.
- Desaturation to <88% or distance <250 m on 6 MWT or 450 m decline in 6 MWT distance over a 6-month period.
- Presence of pulmonary hypertension on right heart catheterization or 2-dimensional echocardiography.
- Hospitalization because of respiratory decline, pneumothorax, or acute exacerbation.
4.5. Pulmonary Rehabilitation
4.6. Palliative Care and Physicological Support
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Suzuki, A.; Kondoh, Y. The clinical impact of major comorbidities on idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir. J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Collard, H.R. Comorbid Conditions in Idiopathic Pulmonary Fibrosis: Recognition and Management. Front. Med. 2017, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Zisman, D.A.; Schwarz, M.; Anstrom, K.J.; Collard, H.R.; Flaherty, K.R.; Hunninghake, G.W. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N. Engl. J. Med. 2010, 363, 620–628. [Google Scholar] [PubMed]
- Hassoun, P.M.; Nathan, S.D. Sildenafil for pulmonary hypertension complicating idiopathic pulmonary fibrosis: A rationale grounded in basic science. Eur. Respir. J. 2016, 47, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.Y.; Jiang, X.; Jing, Z.C. Riociguat: A soluble guanylate cyclase stimulator for the treatment of pulmonary hypertension. Drug Des. Dev. Ther. 2017, 11, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Dong, F.; Song, Q.; Chi, F.; Liu, L.; Wang, Y.; Che, C. Combined pulmonary fibrosis and emphysema: A retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm. Med. 2016, 16, 137. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Wells, A.U.; Nicholson, A.G.; Richeldi, L.; Flaherty, K.R.; Le Maulf, F.; Stowasser, S.; Schlenker-Herceg, R.; Hansell, D.M. Effect of nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. Am. J. Respir. Crit. Care Med. 2017, 195, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.H.; Mason, W.R.; Parnell, J.A.; Rice, T.W.; Loyd, J.E.; Milstone, A.P.; Collard, H.R.; Malow, B.A. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest 2009, 136, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Bosi, M.; Milioli, G.; Fanfulla, F.; Tomassetti, S.; Ryu, J.H.; Parrino, L.; Riccardi, S.; Melpignano, A.; Vaudano, A.E.; Ravaglia, C.; et al. OSA and Prolonged oxygen desaturation during sleep are strong predictors of poor outcome in IPF. Lung 2017, 195, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Mermigkis, C.; Bouloukaki, I.; Schiza, S.E. Sleep as a new target for improving outcomes in idiopathic pulmonary fibrosis. Chest 2017, 152, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collard, H.R.; Anstrom, K.J.; Martinez, F.J.; Noth, I.; Roberts, R.S.; Yow, E.; Raghu, G. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: An analysis of data from three randomised controlled trials. Lancet Respir. Med. 2013, 1, 369–376. [Google Scholar] [CrossRef]
- Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Noth, I.; Anstrom, K.J.; Calvert, S.B.; de Andrade, J.; Flaherty, K.R.; Glazer, C.; Kaner, R.J.; Olman, M.A. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 88–95. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet (Lond.) 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Kreuter, M.; Wuyts, W.; Renzoni, E.; Koschel, D.; Maher, T.M.; Kolb, M.; Weycker, D.; Spagnolo, P.; Kirchgaessler, K.U.; Herth, F.J.; et al. Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: A pooled analysis. Lancet Respir. Med. 2016, 4, 381–389. [Google Scholar] [CrossRef]
- Johannson, K.A.; Strambu, I.; Ravaglia, C.; Grutters, J.C.; Valenzuela, C.; Mogulkoc, N.; Luppi, F.; Richeldi, L.; Wells, A.U.; Vancheri, C.; et al. Antacid therapy in idiopathic pulmonary fibrosis: More questions than answers? Lancet Respir. Med. 2017, 5, 591–598. [Google Scholar] [CrossRef]
- Raghu, G.; Morrow, E.; Collins, B.F.; Ho, L.A.; Hinojosa, M.W.; Hayes, J.M.; Spada, C.A.; Oelschlager, B.; Li, C.; Yow, E.; et al. Laparoscopic anti-reflux surgery for idiopathic pulmonary fibrosis at a single centre. Eur. Respir. J. 2016, 48, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Fidler, L.; Sitzer, N.; Shapera, S.; Shah, P.S. Treatment of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis: A Systematic review and meta-analysis. Chest 2018, 153, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Basavaraj, A.; Reichner, C.; Shlobin, O.A.; Ahmad, S.; Kiernan, J.; Burton, N.; Barnett, S.D. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir. Med. 2010, 104, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.; Nurmi, H.; Kettunen, H.P.; Selander, T.; Purokivi, M.; Kaarteenaho, R. Underlying and immediate causes of death in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2018, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Verma, I.; Shah, V.; Agarwal, A.; Sikachi, R.R. Cardiac manifestations of idiopathic pulmonary fibrosis. Intractable Rare Dis. Res. 2016, 5, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Lever, V.; Rossi, A.; Gilioli, E.; Brunelli, M.; Dubini, A.; Tomassetti, S.; Piciucchi, S.; Nottegar, A.; Rossi, G.; et al. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology 2017, 71, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Watanabe, A.; Kondo, H.; Kanzaki, M.; Okubo, K.; Yokoi, K.; Matsumoto, K.; Marutsuka, T.; Shinohara, H.; Teramukai, S.; et al. Long-term results and predictors of survival after surgical resection of patients with lung cancer and interstitial lung diseases. J. Thorac. Cardiovasc. Surg. 2015, 149, 64–70.e2. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, N.; Tadokoro, A.; Kita, N.; Murota, M.; Ishii, T.; Takagi, T.; Watanabe, N.; Tojo, Y.; Harada, S.; Hasui, Y.; et al. Impact of idiopathic pulmonary fibrosis on advanced non-small cell lung cancer survival. J. Cancer Res. Clin. Oncol. 2016, 142, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Yoshino, I.; Yoshida, S.; Ikeda, N.; Tsuboi, M.; Asato, Y.; Katakami, N.; Sakamoto, K.; Yamashita, Y.; Okami, J.; et al. A phase II trial evaluating the efficacy and safety of perioperative pirfenidone for prevention of acute exacerbation of idiopathic pulmonary fibrosis in lung cancer patients undergoing pulmonary resection: West Japan Oncology Group 6711 L (PEOPLE Study). Respir. Res. 2016, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, S.; Sato, A.; Hayakawa, H.; Kotani, M.; Urano, T.; Takada, A. Tissue factor expression and fibrin deposition in the lungs of patients with idiopathic pulmonary fibrosis and systemic sclerosis. Am. J. Respir. Crit. Care Med. 1997, 156, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.B.; Smith, C.; Le Jeune, I.; Gribbin, J.; Fogarty, A.W. The association between idiopathic pulmonary fibrosis and vascular disease: A population-based study. Am. J. Respir. Crit. Care Med. 2008, 178, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Sode, B.F.; Dahl, M.; Nielsen, S.F.; Nordestgaard, B.G. Venous thromboembolism and risk of idiopathic interstitial pneumonia: A nationwide study. Am. J. Respir. Crit. Care Med. 2010, 181, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Sprunger, D.B.; Olson, A.L.; Huie, T.J.; Fernandez-Perez, E.R.; Fischer, A.; Solomon, J.J.; Brown, K.K.; Swigris, J.J. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur. Respir. J. 2012, 39, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ward, A.J.; Lanes, S.; Cortney Hayflinger, D.; Rosenberg, D.M.; Hunsche, E. Burden of illness in idiopathic pulmonary fibrosis. J. Med. Econ. 2012, 15, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, G.; Wenter, V.; Milger, K.; Zimmermann, G.S.; Matthes, S.; Meinel, F.G.; Lehner, S.; Neurohr, C.; Behr, J.; Kneidinger, N. Suspected pulmonary embolism in patients with pulmonary fibrosis: Discordance between ventilation/perfusion SPECT and CT pulmonary angiography. Respirology (Carlton) 2016, 21, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.V.; Barco, S.; Lankeit, M.; Meyer, G. Management of pulmonary embolism: An update. J. Am. Coll. Cardiol. 2016, 67, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.V.; Torbicki, A. Management of pulmonary embolism: Recent evidence and the new European guidelines. Eur. Respir. J. 2014, 44, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Usuki, J.; Azuma, A.; Nakagawa, T.; Kudoh, S. Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest 2003, 123, 2007–2011. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, Z.; Yu, X.; Zhu, H.; Qiu, W.; Fan, L.; Yi, X. Diabetes mellitus in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2016, 9, 17727–17738. [Google Scholar]
- Garber, J.R.; Cobin, R.H.; Gharib, H.; Hennessey, J.V.; Klein, I.; Mechanick, J.I.; Pessah-Pollack, R.; Singer, P.A.; Woeber, K.A. Clinical practice guidelines for hypothyroidism in adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2012, 18, 988–1028. [Google Scholar]
- Oldham, J.M.; Kumar, D.; Lee, C.; Patel, S.B.; Takahashi-Manns, S.; Demchuk, C.; Strek, M.E.; Noth, I. Thyroid Disease Is Prevalent and Predicts Survival in Patients With Idiopathic Pulmonary Fibrosis. Chest 2015, 148, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, C.J.; Berkeley, J.; Carrieri-Kohlman, V.L.; Pantilat, S.Z.; Landefeld, C.S.; Collard, H.R. Depression and functional status are strongly associated with dyspnea in interstitial lung disease. Chest 2011, 139, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.E.; Fiore, J.F., Jr.; Bell, E.C.; Goh, N.; Westall, G.; Symons, K.; Dowman, L.; Glaspole, I. Dyspnoea and comorbidity contribute to anxiety and depression in interstitial lung disease. Respirology (Carlton) 2014, 19, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Glaspole, I.N.; Watson, A.L.; Allan, H.; Chapman, S.; Cooper, W.A.; Corte, T.J.; Ellis, S.; Grainge, C.; Goh, N.; Hopkins, P.; et al. Determinants and outcomes of prolonged anxiety and depression in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Usmani, Z.A.; Carson, K.V.; Heslop, K.; Esterman, A.J.; De Soyza, A.; Smith, B.J. Psychological therapies for the treatment of anxiety disorders in chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2017, 3, Cd010673. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Cottin, V.; Brown, K. Recent lessons learned in the management of acute exacerbations of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 170050. [Google Scholar] [CrossRef] [PubMed]
- Papiris, S.A.; Kagouridis, K.; Kolilekas, L.; Papaioannou, A.I.; Roussou, A.; Triantafillidou, C.; Baou, K.; Malagari, K.; Argentos, S.; Kotanidou, A.; et al. Survival in Idiopathic pulmonary fibrosis acute exacerbations: The non-steroid approach. BMC Pulm. Med. 2015, 15, 162. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Richeldi, L.; Kim, D.S.; Taniguchi, H.; Tschoepe, I.; Luisetti, M.; Roman, J.; Tino, G.; Schlenker-Herceg, R.; Hallmann, C.; et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Tsushima, K.; Abe, M.; Suzuki, K.; Yamagishi, K.; Matsumura, A.; Ichimura, Y.; Ikari, J.; Terada, J.; Tatsumi, K. The effects of pirfenidone in patients with an acute exacerbation of interstitial pneumonia. Clin. Respir. J. 2018, 12, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Costabel, U.; Richeldi, L.; Cottin, V.; Wijsenbeek, M.; Bonella, F.; Bendstrup, E.; Maher, T.M.; Wachtlin, D.; Stowasser, S.; et al. Statin Therapy and Outcomes in Trials of Nintedanib in Idiopathic Pulmonary Fibrosis. Respir. Int. Rev. Thorac. Dis. 2018, 95, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Douglas, W.W.; Ryu, J.H.; Schroeder, D.R. Idiopathic pulmonary fibrosis: Impact of oxygen and colchicine, prednisone, or no therapy on survival. Am. J. Respir. Crit. Care Med. 2000, 161, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.C.; Cox, N.S.; Goh, N.; Glaspole, I.; Westall, G.P.; Watson, A.; Holland, A.E. Oxygen therapy for interstitial lung disease: A systematic review. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.J. Follow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patient. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2011, 20, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Visca, D.; Montgomery, A.; de Lauretis, A.; Sestini, P.; Soteriou, H.; Maher, T.M.; Wells, A.U.; Renzoni, E.A. Ambulatory oxygen in interstitial lung disease. Eur. Respir. J. 2011, 38, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Hallstrand, T.S.; Boitano, L.J.; Johnson, W.C.; Spada, C.A.; Hayes, J.G.; Raghu, G. The timed walk test as a measure of severity and survival in idiopathic pulmonary fibrosis. Eur. Respir. J. 2005, 25, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Andrei, A.C.; Murray, S.; Fraley, C.; Colby, T.V.; Travis, W.D.; Lama, V.; Kazerooni, E.A.; Gross, B.H.; Toews, G.B.; et al. Idiopathic pulmonary fibrosis: Prognostic value of changes in physiology and six-minute-walk test. Am. J. Respir. Crit. Care Med. 2006, 174, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, C.J.; Nathan, S.D.; Browning, R.F.; Barnett, S.D.; Ahmad, S.; Shorr, A.F. The distance-saturation product predicts mortality in idiopathic pulmonary fibrosis. Respir. Med. 2006, 100, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Hook, J.L.; Arcasoy, S.M.; Zemmel, D.; Bartels, M.N.; Kawut, S.M.; Lederer, D.J. Titrated oxygen requirement and prognostication in idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: A clinical trial. Ann. Intern. Med. 1980, 93, 391–398. [Google Scholar]
- Medical Research Council Working Party. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Lancet 1981, 1, 681–686. [Google Scholar]
- Hardinge, M.; Annandale, J.; Bourne, S.; Cooper, B.; Evans, A.; Freeman, D.; Green, A.; Hippolyte, S.; Knowles, V.; MacNee, W.; et al. British Thoracic Society guidelines for home oxygen use in adults. Thorax 2015, 70 (Suppl. 1), i1–i43. [Google Scholar] [CrossRef] [PubMed]
- Faverio, P.; De Giacomi, F.; Sardella, L.; Fiorentino, G.; Carone, M.; Salerno, F.; Ora, J.; Rogliani, P.; Pellegrino, G.; Sferrazza Papa, G.F.; et al. Management of acute respiratory failure in interstitial lung diseases: Overview and clinical insights. BMC Pulm. Med. 2018, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Gaudry, S.; Vincent, F.; Rabbat, A.; Nunes, H.; Crestani, B.; Naccache, J.M.; Wolff, M.; Thabut, G.; Valeyre, D.; Cohen, Y.; et al. Invasive mechanical ventilation in patients with fibrosing interstitial pneumonia. J. Thorac. Cardiovasc. Surg. 2014, 147, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Vianello, A.; Pipitone, E. Noninvasive ventilation in patients with idiopathic pulmonary fibrosis is not a futile intervention! J. Crit. Care 2014, 29, 1129. [Google Scholar] [CrossRef] [PubMed]
- Gungor, G.; Tatar, D.; Salturk, C.; Cimen, P.; Karakurt, Z.; Kirakli, C.; Adiguzel, N.; Ediboglu, O.; Yilmaz, H.; Mocin, O.Y.; et al. Why do patients with interstitial lung diseases fail in the ICU? A 2-center cohort study. Respir. Care 2013, 58, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Rozencwajg, S.; Schmidt, M. Extracorporeal membrane oxygenation for interstitial lung disease: What is on the other side of the bridge? J. Thorac. Dis. 2016, 8, 1918–1920. [Google Scholar] [CrossRef] [PubMed]
- Trudzinski, F.C.; Kaestner, F.; Schafers, H.J.; Fahndrich, S.; Seiler, F.; Bohmer, P.; Linn, O.; Kaiser, R.; Haake, H.; Langer, F.; et al. Outcome of patients with interstitial lung disease treated with extracorporeal membrane oxygenation for acute respiratory failure. Am. J. Respir. Crit. Care Med. 2016, 193, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Fuehner, T.; Kuehn, C.; Hadem, J.; Wiesner, O.; Gottlieb, J.; Tudorache, I.; Olsson, K.M.; Greer, M.; Sommer, W.; Welte, T.; et al. Extracorporeal membrane oxygenation in awake patients as bridge to lung transplantation. Am. J. Respir. Crit. Care Med. 2012, 185, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kistler, K.D.; Nalysnyk, L.; Rotella, P.; Esser, D. Lung transplantation in idiopathic pulmonary fibrosis: A systematic review of the literature. BMC Pulm. Med. 2014, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Chambers, D.C.; Yusen, R.D.; Cherikh, W.S.; Goldfarb, S.B.; Kucheryavaya, A.Y.; Khusch, K.; Levvey, B.J.; Lund, L.H.; Meiser, B.; Rossano, J.W.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-fourth adult lung and heart-lung transplantation report-2017; Focus Theme: Allograft ischemic time. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2017, 36, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Force, S.D.; Kilgo, P.; Neujahr, D.C.; Pelaez, A.; Pickens, A.; Fernandez, F.G.; Miller, D.L.; Lawrence, C. Bilateral lung transplantation offers better long-term survival, compared with single-lung transplantation, for younger patients with idiopathic pulmonary fibrosis. Ann. Thorac. Surg. 2011, 91, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Gulack, B.C.; Ganapathi, A.M.; Speicher, P.J.; Meza, J.M.; Hirji, S.A.; Snyder, L.D.; Davis, R.D.; Hartwig, M.G. What is the optimal transplant for older patients with idiopathic pulmonary fibrosis? Ann. Thorac. Surg. 2015, 100, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Adamali, H.I.; Anwar, M.S.; Russell, A.-M.; Egan, J.J. Non-pharmacological treatment of idiopathic pulmonary fibrosis. Curr. Respir. Care Rep. 2012, 1, 208–215. [Google Scholar] [CrossRef]
- Rochester, C.L.; Vogiatzis, I.; Holland, A.E.; Lareau, S.C.; Marciniuk, D.D.; Puhan, M.A.; Spruit, M.A.; Masefield, S.; Casaburi, R.; Clini, E.M.; et al. An official American Thoracic Society/European Respiratory Society policy statement: Enhancing implementation, use, and delivery of pulmonary rehabilitation. Am. J. Respir. Crit. Care Med. 2015, 192, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Dowman, L.; Hill, C.J.; Holland, A.E. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst. Rev. 2014, Cd006322. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.E.; Hill, C.J.; Conron, M.; Munro, P.; McDonald, C.F. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax 2008, 63, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, O.; Miyajima, H.; Fukai, Y.; Yamazaki, R.; Satoh, R.; Yamagata, T.; Sano, H.; Iwanaga, T.; Higashimoto, Y.; Nakajima, H.; et al. Effect of ambulatory oxygen on exertional dyspnea in IPF patients without resting hypoxemia. Respir. Med. 2013, 107, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Huppmann, P.; Sczepanski, B.; Boensch, M.; Winterkamp, S.; Schonheit-Kenn, U.; Neurohr, C.; Behr, J.; Kenn, K. Effects of inpatient pulmonary rehabilitation in patients with interstitial lung disease. Eur. Respir. J. 2013, 42, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.M.; Gomez-Marin, O.W.; Ramos, C.F.; Sol, C.M.; Cohen, M.I.; Gaunaurd, I.A.; Cahalin, L.P.; Cardenas, D.D. Exercise limitation in IPF patients: A randomized trial of pulmonary rehabilitation. Lung 2014, 192, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Batiste, X.; Espinosa, J.; Porta-Sales, J.; Benito, E. Models of care, organization and quality improvement for the care of advanced and terminal patients and their families: The contribution of palliative care. Med. Clin. 2010, 135, 83–89. [Google Scholar]
- Kreuter, M.; Bendstrup, E.; Russell, A.M.; Bajwah, S.; Lindell, K.; Adir, Y.; Brown, C.E.; Calligaro, G.; Cassidy, N.; Corte, T.J.; et al. Palliative care in interstitial lung disease: Living well. Lancet Respir. Med. 2017, 5, 968–980. [Google Scholar] [CrossRef]
- Kalluri, M.; Claveria, F.; Ainsley, E.; Haggag, M.; Armijo-Olivo, S.; Richman-Eisenstat, J. Beyond idiopathic pulmonary fibrosis diagnosis: Multidisciplinary care with an early integrated palliative approach is associated with a decrease in acute care utilization and hospital deaths. J. Pain Symptom Manag. 2018, 55, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Lindell, K.O.; Liang, Z.; Hoffman, L.A.; Rosenzweig, M.Q.; Saul, M.I.; Pilewski, J.M.; Gibson, K.F.; Kaminski, N. Palliative care and location of death in decedents with idiopathic pulmonary fibrosis. Chest 2015, 147, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Hoffman, L.A.; Nouraie, M.; Kass, D.J.; Donahoe, M.P.; Gibson, K.F.; Saul, M.I.; Lindell, K.O. Referral to palliative care infrequent in patients with idiopathic pulmonary fibrosis admitted to an intensive care unit. J. Palliat. Med. 2017, 20, 134–140. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millan-Billi, P.; Serra, C.; Alonso Leon, A.; Castillo, D. Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 59. https://doi.org/10.3390/medsci6030059
Millan-Billi P, Serra C, Alonso Leon A, Castillo D. Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis. Medical Sciences. 2018; 6(3):59. https://doi.org/10.3390/medsci6030059
Chicago/Turabian StyleMillan-Billi, Paloma, Candela Serra, Ana Alonso Leon, and Diego Castillo. 2018. "Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis" Medical Sciences 6, no. 3: 59. https://doi.org/10.3390/medsci6030059
APA StyleMillan-Billi, P., Serra, C., Alonso Leon, A., & Castillo, D. (2018). Comorbidities, Complications and Non-Pharmacologic Treatment in Idiopathic Pulmonary Fibrosis. Medical Sciences, 6(3), 59. https://doi.org/10.3390/medsci6030059