Apoptosis: Activation and Inhibition in Health and Disease

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
2. Generalities of Apoptosis
3. Activation
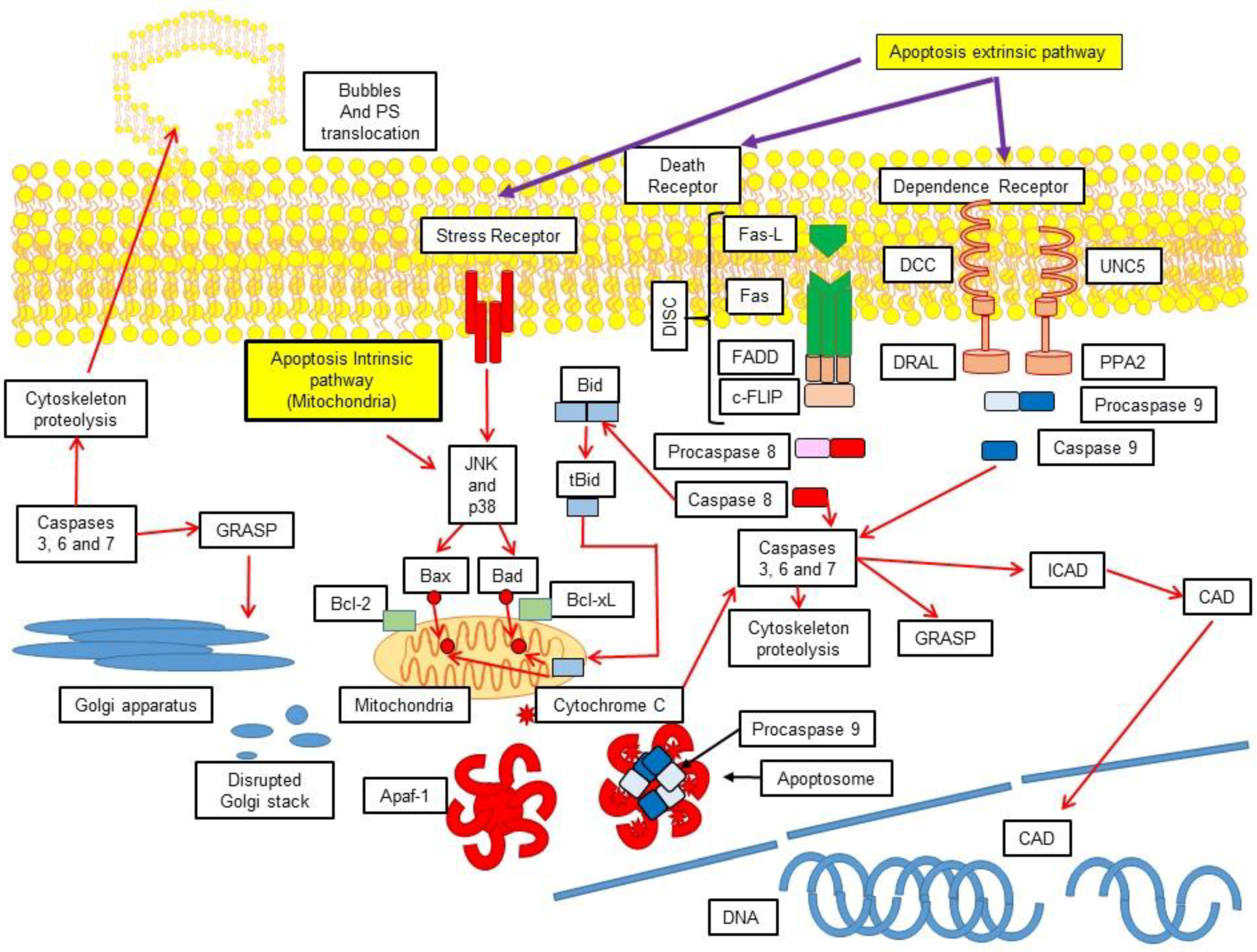
3.1. Extrinsic Pathway
3.2. Activation of the Intrinsic Pathway via the Mitochondrial-Induced Apoptosis
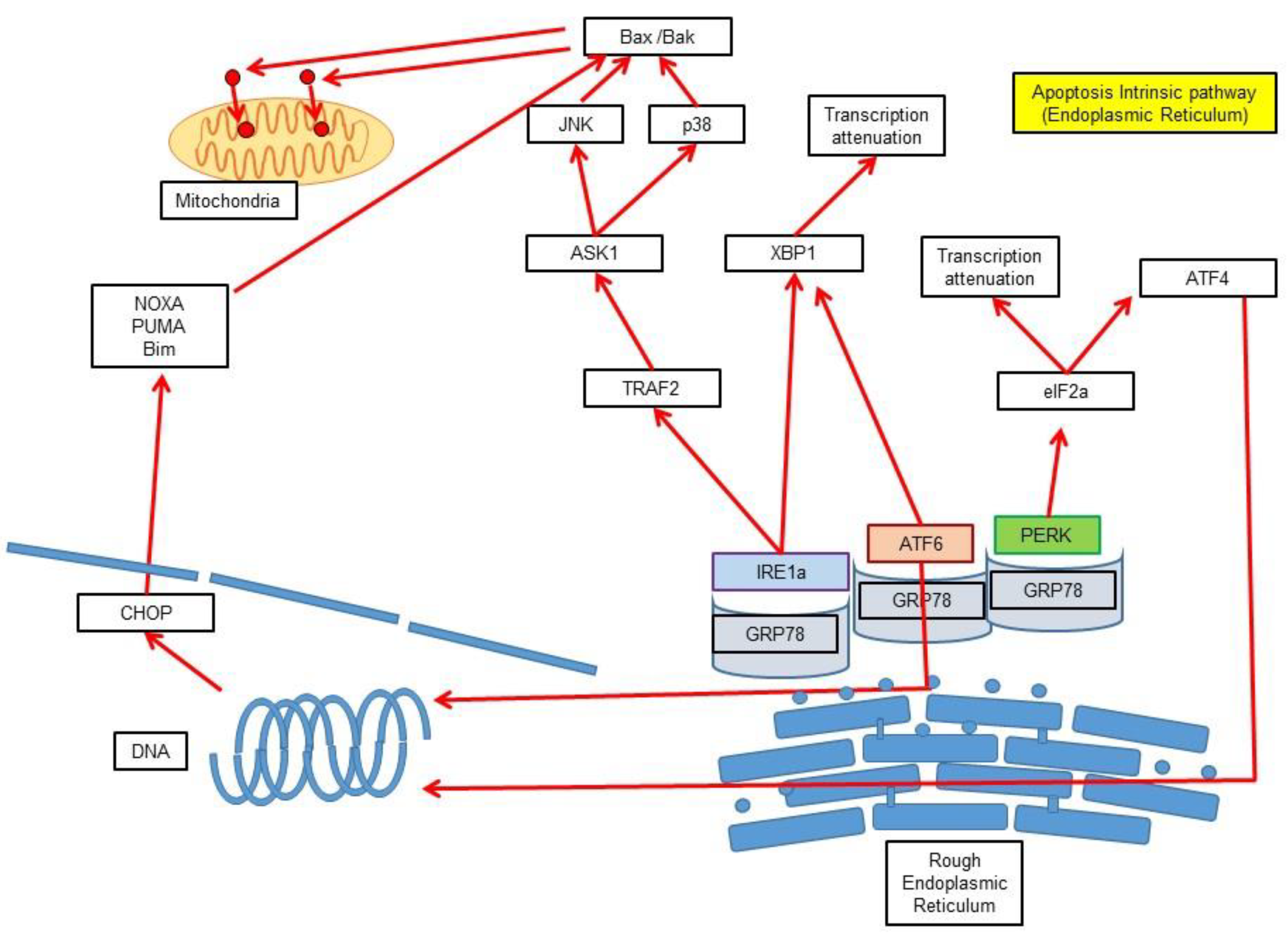
3.3. Intrinsic Pathway via Endoplasmic Reticulum Stress-Induced Apoptosis
3.4. Caspase Independent Pathway
4. Execution
4.1. Generalities of Caspases
4.2. Formation of the Apoptosome
5. Cellular Demolition
6. Signal Transduction Pathways in Apoptosis
6.1. Mitogen-Activated Protein Kinase (MAPK) Family
6.2. p38
6.3. Jun N-Terminal Kinase (JNK)
6.4. Extracellular Signal–Regulated Kinase 1/2 (ERK1/2)
7. Participation of MAPK in Apoptosis
8. Transforming Growth Factor-β-Activated Kinase 1 (TAK1) and Apoptosis
9. Phosphatidylinositol 3-Kinase (PI3K)/Akt Signalling Pathway
10. Apoptosis in Physiological Processes
11. Apoptosis Inhibition and Infection
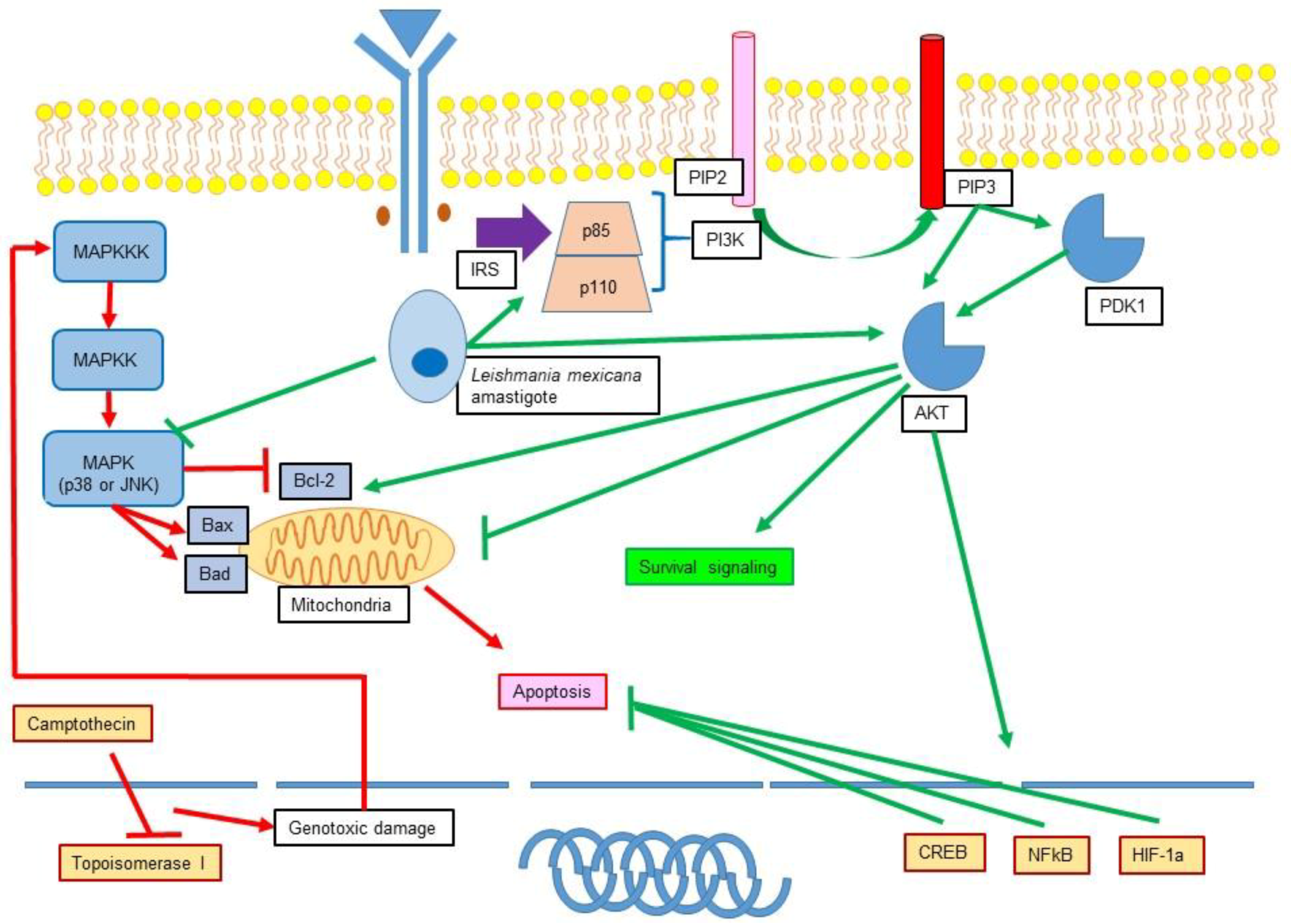
11.1. Leishmania Participation in Apoptosis Inhibition
11.2. Role of Leishmania in the Modulation of MAPK and PI3K
12. Apoptosis Inhibition and Cancer
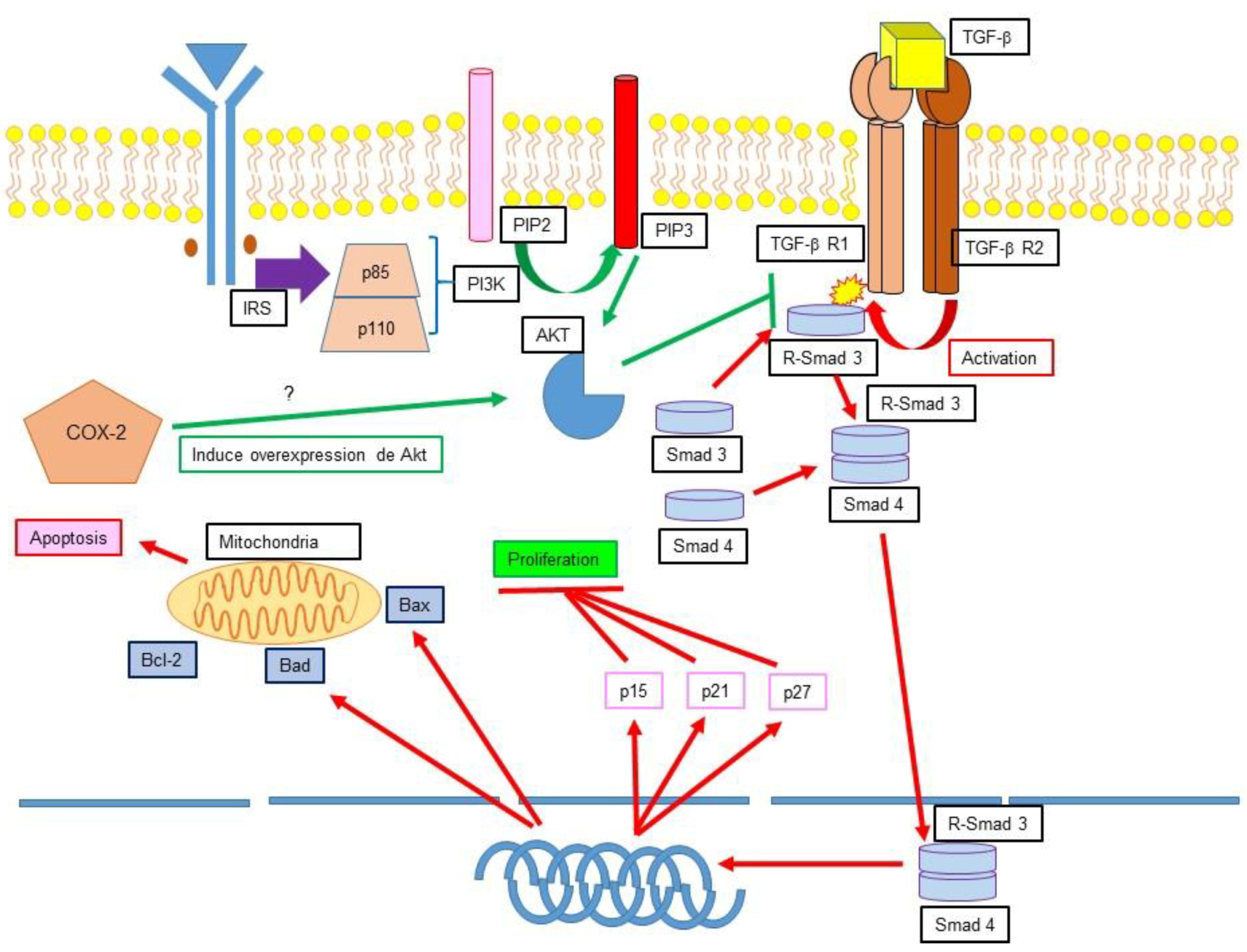
12.1. TGF-β, Apoptosis Inhibition and Cancer
12.2. Ciclooxygenase 2 (COX-2) Apoptosis Inhibition and Cancer
13. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2008, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2011, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signalling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Budd, R.C.; Desbarats, J.; Hedrick, S.M.; Hueber, A.O.; Newell, M.K.; Owen, L.B.; Pope, R.M.; Tschopp, J.; Wajant, H.; et al. The CD95 receptor: Apoptosis revisited. Cell 2007, 129, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Fas signalling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Schutze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Bredesen, D.E. Dependence receptors: From basic research to drug development. Sci. Signal. 2011. [Google Scholar] [CrossRef] [PubMed]
- Koshio, O.; Nagao, T.; Mabuchi, A.; Ono, Y.; Suzuki, K. Apoptotic signalling in endothelial cells with neutrophil activation. Mol. Cell. Biochem. 2012, 363, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Janssen, W.J.; Barthel, L.; Muldrow, A.; Oberley-Deegan, R.E.; Kearns, M.T.; Jakubzick, C.; Henson, P.M. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2011, 184, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schröter, M.; et al. Viral FLICE inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006, 6, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, A novel FADD-homologous ICE/CED-3–like protease, is recruited to the CD95 (Fas/APO-1) death-Inducing signalling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef]
- Meier, P.; Vousden, K.H. Lucifer’s labyrinth—Ten years of path finding in cell death. Mol. Cell 2007, 28, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I. Death receptor signalling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signalling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signalling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [PubMed]
- Guenebeaud, C.; Goldschneider, D.; Castets, M.; Guix, C.; Chazot, G.; Delloye-Bourgeois, C.; Eisenberg-Lerner, A.; Shohat, G.; Zhang, M.; Laudet, V.; et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol. Cell 2010, 40, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Grespi, F.; Chmelewskij, W.; Villunger, A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis 2009, 14, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.L. From mitochondrial biology to magic bullet: Navitoclax disarms BCL-2 in chronic lymphocytic leukemia. J. Clin. Oncol. 2012, 30, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef] [PubMed]
- Vařecha, M.M.; Potěšilová, M.M.; Matula, P.P.; Kozubek, M.M. Endonuclease G interacts with histone H2B and DNA topoisomerase II alpha during apoptosis. Mol. Cell. Biochem. 2012, 363, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Damle, S.; Jugdutt, B.I.; Moe, G.W. Nuclear-mitochondrial cross-talk in global myocardial ischemia. A time-course analysis. Mol. Cell. Biochem. 2012, 364, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010, 16, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Shi, Y. Apoptosome and inflammasome: Conserved machineries for caspase activation. Natl. Sci. Rev. 2014, 1, 101–118. [Google Scholar] [CrossRef]
- Mcluskey, K.; Mottram, J.C. Comparative structural analysis of the caspase family with other clan CD cysteine peptidases. Biochem. J. 2015, 466, 219–232. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.H.; Clarck, A.C. Death by caspase dimerization. Adv. Exp. Med. Biol. 2012, 747, 55–73. [Google Scholar] [PubMed]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Lu, W.; Rabinowitz, J.D.; Shi, Y. Calcium blocks formation of apoptosome by preventing nucleotide exchange in Apaf-1. Mol. Cell 2007, 25, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Qin, H.; Srinivasula, S.M.; Wu, G.; Fernandes-Alnemri, T.; Alnemri, E.S.; Shi, Y. Structural basis of procaspase-9 recruitment by the apoptotic protease activating factor 1. Nature 1999, 399, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Sumandea, M.; Tombe, P.D.; Narula, J.; Solaro, R.J.; Hajjar, R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 6252–6256. [Google Scholar] [CrossRef] [PubMed]
- Gerner, C.; Fröhwein, U.; Gotzmann, J.; Bayer, E.; Gelbmann, D.; Bursch, W.; Schulte-Hermann, R. The fas-induced apoptosis analyzed by high throughput proteome analysis. J. Biol. Chem. 2000, 275, 39018–39026. [Google Scholar] [CrossRef] [PubMed]
- O’brien, G.A. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J. Biol. Chem. 1995, 270, 6425–6428. [Google Scholar]
- Thiede, B.; Treumann, A.; Kretschmer, A.; Sohike, J.; Rudel, T. Shotgun proteome analysis of protein changes in apoptotic cells. Proteomics 2005, 5, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Kothakota, S. Caspase-3-generated fragment of gelsolin: Effector of morphological change in apoptosis. Science 1997, 278, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.A.; Johnstone, R.W.; Jans, D.A.; Trapani, J.A. Filamin (280-kDa actin-binding Protein) is a caspase substrate and is also cleaved directly by the cytotoxic T lymphocyte protease granzyme B during apoptosis. J. Biol. Chem. 2000, 275, 39262–39266. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Duriez, P.J.; Brumatti, G.; Delivani, P.; Martin, S.J. The cytotoxic lymphocyte protease, granzyme B, targets the cytoskeleton and perturbs microtubule polymerization dynamics. J. Biol. Chem. 2006, 281, 8118–8125. [Google Scholar] [CrossRef] [PubMed]
- Canu, N.; Dus, L.; Barbato, C.; Ciotti, M.; Brancolini, C.; Rinaldi, A.; Novak, M.; Cattaneo, A.; Bradbury, A.; Calissano, P. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J. Neurosci. 1998, 18, 7061–7064. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.D.; Vergnolle, M.A.; Woodman, P.G.; Allan, V.J. Apoptotic Cleavage of Cytoplasmic Dynein Intermediate Chain and P150 Glued Stops Dynein-Dependent Membrane Motility. J. Cell Biol. 2001, 153, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N. Changes in nuclear morphology during apoptosis correlate with vimentin cleavage by different caspases located either upstream or downstream of Bcl-2 action. Genes Cells 1999, 4, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.-O.; Liao, J.; Omary, M.B. Apoptosis generates stable fragments of human type I keratins. J. Biol. Chem. 1997, 272, 33197–33203. [Google Scholar] [CrossRef] [PubMed]
- Orth, K.; Chinnaiyan, A.M.; Garg, M.; Froelich, C.J.; Dixit, V.M. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol. Chem. 1996, 271, 16443–16446. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, C.; Passante, E.; Rehm, M. The Molecular Machinery Regulating Apoptosis Signal Transduction and its Implication in Human Physiology and Pathophysiologies. Curr. Mol. Med. 2011, 11, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, A.; Grimm, S. Mitochondrial respiratory chain complexes: Apoptosis sensors mutated in cancer? Oncogene 2011, 30, 3985. [Google Scholar] [CrossRef] [PubMed]
- Mierke, C. Physical view on migration modes. Cell Adhes. Migr. 2015, 9, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, C.; Bergmann, A. The Sound of Silence: Signalling by Apoptotic Cells. Curr. Top. Dev. Biol. 2015, 114, 241–265. [Google Scholar] [PubMed]
- Martinet, W.; De Meyer, I.; Cools, N.; Timmerman, V.; Bult, H.; Bosmans, J.; De Meyer, G.R. Cell Death–Mediated Cleavage of the Attraction Signal p43 in Human Atherosclerosis Implications for Plaque Destabilization. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Obakan-Yerlikaya, P.; Arisan, E.D.; Coker-Gurkan, A.; Adacan, K.; Ozbey, U.; Somuncu, B.; Baran, D.; Palavan-Unsal, N. Calreticulin is a fine tuning molecule in epibrassinolide-induced apoptosis through activating endoplasmic reticulum stress in colon cancer cells. Mol. Carcinog. 2017, 56, 1603–1619. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D.; Whitmarsh, A.J. MAP kinase signalling cascades and transcriptional regulation. Gene 2013, 513, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Krens, S.G.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006, 580, 4984–4990. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Ryazantseva, N.V.; Novitsky, V.V.; Chasovskih, N.Y.; Kaygorodova, E.V.; Starikova, E.G.; Starikov, Y.V.; Radzivil, T.T. Role of recombinant mitogen-activated protein kinases JNK and p38 in the regulation of apoptosis in blood mononuclear cells under conditions of oxidative stress in vitro. Bull. Exp. Biol. Med. 2008, 145, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Sassmann-Schweda, A.; Singh, P.; Tang, C.; Wietelmann, A.; Wettschureck, N.; Offermanns, S. Increased apoptosis and browning of TAK1-deficient adipocytes protects against obesity. JCI Insight 2016, 1, e81175. [Google Scholar] [CrossRef] [PubMed]
- Morioka, S.; Broglie, P.; Omori, E.; Ikeda, Y.; Takaesu, G.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J. Cell Biol. 2014, 204, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Keys, J.R.; Strickler, J.E.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Loesch, M. The p38 MAPK stress pathway as a tumor suppressor or more? Front. Biosci. 2008, 13, 3581–3593. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signalling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Waskiewicz, A.J.; Cooper, J.A. Mitogen and stress response pathways: MAP kinase cascades and phosphatase regulation in mammals and yeast. Curr. Opin. Cell Biol. 1995, 7, 798–805. [Google Scholar] [CrossRef]
- Tibbles, L.A.; Woodgett, J.R. The stress-activated protein kinase pathways. Cell. Mol. Life Sci. 1999, 55, 1230–1254. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Bulvin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J., Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Han, J. The p38 signal transduction pathway activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Sun, W.; Qu, B.; Cardona, C.J.; Powell, K.; Wegner, M.; Shi, Y.; Xing, Z. Distinct regulation of host responses by ERK and JNK MAP kinases in swine macrophages infected with pandemic (H1N1) 2009 influenza virus. PLoS ONE 2012, 7, e30328. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Koepke, A.; Jarad, G.; Schlessman, K.; Cleveland, R.P.; Wang, B.; Konieczkowski, M.; Schelling, J.R. Apoptosis and JNK activation are differentially regulated by Fas expression level in renal tubular epithelial cells. Kidney Int. 2001, 60, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Zhao, X.; Im, J.Y.; Grosso, H.; Jang, W.H.; Chan, T.W.; Sonsalla, P.K.; German, D.C.; Ichijo, H.; Junn, E.; et al. Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS ONE 2012, 7, e29935. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK cascade. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- Dérijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Su, B.; Tsigelny, I.; Sluss, H.K.; Dérijard, B.; Moore, G.; Davis, R.; Karin, M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994, 8, 2996–3007. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [PubMed]
- Dreskin, S.C.; Thomas, G.W.; Dale, S.N.; Heasley, L.E. Isoforms of jun kinase are differentially expressed and activated in human monocyte/macrophage (THP-1) cells. J. Immunol. 2001, 166, 5646–5653. [Google Scholar] [CrossRef] [PubMed]
- Kumagae, Y.; Zhang, Y.; Kim, O.-J.; Miller, C.A. Human c-Jun N-terminal kinase expression and activation in the nervous system. Mol. Brain Res. 1999, 67, 10–17. [Google Scholar] [CrossRef]
- Chen, F.; Beezhold, K.; Castranova, V. JNK1, a potential therapeutic target for hepatocellular carcinoma. Biochim. Biophys. Acta 2009, 1796, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Tafolla, E. JNK1 and JNK2 Oppositely regulate p53 in signalling linked to apoptosis triggered by an altered fibronectin matrix: JNK links FAK and p53. J. Biol. Chem. 2005, 280, 19992–19999. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.-T.; Qiu, W.R.; Wang, Y.-P. JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene 1997, 15, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Tu, H.C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.D.; Cheng, E.H.Y. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Werlen, G. Signalling life and death in the thymus: Timing is everything. Science 2003, 299, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Chang, S.H.; Becker, E.B.E.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Masot, X.A.; Hetman, M.; Higgins, M.J.; Kokot, N.; Xia, Z. Taxol induces apoptosis in cortical neurons by a mechanism independent of Bcl-2 phosphorylation. J. Neurosci. 2001, 21, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Porras, A. p38 mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol. Biol. Cell 2003, 15, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, S.; Álvarez-Barrientos, A.; Gutiérrez-Uzquiza, A.; Benito, M.; Nebreda, A.R.; Porras, A. Negative regulation of Akt activity by p38α MAP kinase in cardiomyocytes involves membrane localization of PP2A through interaction with caveolin-1. Cell. Signal. 2007, 19, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Bohsali, A.; Abdalla, H.; Velmurugan, K.; Briken, V. The non-pathogenic mycobacteria M. smegmatis and M. fortuitum induce rapid host cell apoptosis via a caspase-3 and TNF dependent pathway. BMC Microbiol. 2010, 10, 237–248. [Google Scholar] [PubMed]
- Zaru, R.; Ronkina, N.; Gaestel, M.; Arthur, J.S.; Watts, C. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signalling response in dendritic cells and is activated through two distinct pathways. Nat. Immunol. 2007, 8, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L. Important aspects of Toll-like receptors, ligands and their signalling pathways. Inflamm. Res. 2010, 59, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Granucci, F. Regulation of antigen uptake, migration, and lifespan of dendritic cell by Toll-like receptors. J. Mol. Med. 2010, 88, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lee, S.W.; Sung, Y.C. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-oh kinase pathway. J. Immunol. 2002, 168, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Sun, H.; Kerfant, B.G.; Crackower, M.A.; Penninger, J.M.; Backx, P.H. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 2004, 37, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [PubMed]
- Bellacosa, A.T.; Staal, S.; Tsichlis, P. A retroviral oncogene, Akt, encoding a serine threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.K.; Ellis, C.A.; Edwards, P.A.; Hiles, I.D. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene 2000, 18, 8024–8032. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef]
- Chen, X.; Thakkar, H.; Tyan, F.; Gim, S.; Robinson, H.; Lee, C.; Pandey, S.K.; Nwokorie, C.; Onwudiwe, N.; Srivastava, R.K. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene 2001, 20, 6073–6083. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, X.; Hernandez, A.; Hellmich, M.R.; Gatalica, Z.; Evers, B.M. Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J. Biol. Chem. 2002, 277, 36602–36610. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Filiz, G.; Brown, D.V.; Hollande, F.; Gonzales, M.; D’Abaco, G.; Papalexis, N.; Phillips, W.A.; Malaterre, J.; Ramsay, R.G.; et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis 2014, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Kuang, C.Y.; Zhu, J.K.; Yu, Y.; Qin, Z.X.; Liu, J.; Huang, L. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol. Cell. Biochem. 2012, 363, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Agani, F.; Jiang, B.-H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Schinoni, M.; Paraná, R. Circulating and Hepatic Fas Expression in HCV-Induced Chronic Liver Disease and Hepatocellular Carcinoma. Gastroenterol. Latinoam. 2006, 36, 211–217. [Google Scholar]
- Norris, D. Differential Control of Cell Death in the Skin. Arch. Dermatol. 1995, 131, 945–948. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.; Cai, J.; Jones, D. Diet and Apoptosis. Ann. Rev. Nutr. 2000, 20, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.; Whang, K.; David-Bajar, K.; Bennion, D. The influence of Ultraviolet Light on Immnunological Cytotoxicity in the Skin. Photochem. Photobiol. 1997, 65, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, L. Electron microscopic observations of duskeratosis, apoptosis, colloid bodies and fibrillar degeneration after skin irritation with dithranol. J. Cutan. Pathol. 1990, 17, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Astals, M.; Laborda, R.M.; Casanova, J.M. Apoptosis y Piel. Dermatol. Venez. 1996, 11, 242–251. [Google Scholar]
- Markstrom, E.; Svensson, E.C.; Shao, R.; Svanberg, B.; Billig, H. Survival factors regulating ovarian apoptosis—Dependence on follicle differentiation. Reproduction 2002, 123, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sugino, N.; Suzuki, T.; Kashida, S.; Karube, A.; Takiguchi, S.; Kato, H. Expression of Bcl-2 and Bax in the human corpus luteum during the menstrual cycle and in early pregnancy: Regulation by human chorionic gonadotropin. J. Clin. Endocrinol. Metab. 2000, 85, 4379–4386. [Google Scholar] [PubMed]
- Gavrilescu, L.C.; Denkers, E.Y. Apoptosis and the Balance of Homeostatic and Pathologic Responses to Protozoan Infection. Infect. Immun. 2003, 71, 6109–6115. [Google Scholar] [CrossRef] [PubMed]
- Terrazas, C.A.; Terrazas, L.I.; Gómez-García, L. Modulation of dendritic cell responses by parasites: A common strategy to survive. J. Biomed. Biotechnol. 2010, 2010, 357106. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowski, S.; Sajid, M.; Reece, S.E. Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasites Vectors 2011, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Matlashewski, G. Intracellular infection by Leishmania donovani inhibits macrophage apoptosis. J. Immunol. 1994, 152, 2930–2937. [Google Scholar] [PubMed]
- Akarid, K.; Arnoult, D.; Micic-Polianski, J.; Sif, J.; Estaquier, J.; Ameisen, J.C. Leishmania major-mediated prevention of programmed cell death induction in infected macrophages is associated with the repression of mitochondrial release of cytochrome c. J. Leukoc. Biol. 2004, 76, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Acquafredda, A.; Spinelli, R.; Schiavone, M.A.; Mitolo, V.; Brandonisio, O.; Panaro, M.A. Infection with Leishmania infantum Inhibits actinomycin d-Induced apoptosis of human monocytic cell line U-937. J. Eukaryot. Microbiol. 2005, 52, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.J.M.; Maciuba, B.Z.B.; Mahan, C.E.C.; McDowell, M.A.M. Leishmania infection inhibits cycloheximide-induced macrophage apoptosis in a strain-dependent manner. Exp. Parasitol. 2009, 123, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Aga, E.; Katschinski, D.M.; van Zandbergen, G.; Laufs, H.; Hansen, B.; Müller, K.; Solbach, W.; Laskay, T. Inhibition of the spontaneous apoptosis of neutrophil granulocytes by the intracellular parasite Leishmania major. J. Immunol. 2002, 169, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Reyes, L.; Argueta, J.; Morán, J.; Salaiza, N.; Hernández, J.; Berzunza, M.; Aguirre-García, M.; Becker, I.; Gutiérrez-Kobeh, L. Leishmania mexicana: Inhibition of camptothecin-induced apoptosis of monocyte-derived dendritic cells. Exp. Parasitol. 2009, 121, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Kobeh, L.; De Oyarzabal, E.; Argueta, J.; Wilkins, A.; Salaiza, N.; Fernández, E.; López, O.; Aguirre, M.; Becker, I. Inhibition of dendritic cell apoptosis by Leishmania mexicana amastigotes. J. Parasitol. Res. 2013, 112, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-González, J.; Wilkins-Rodríguez, A.; Argueta-Donohué, J.; Aguirre-García, M.; Gutiérrez-Kobeh, L. Leishmania mexicana promastigotes down regulate JNK and p-38 MAPK activation: Role in the inhibition of camptothecin-induced apoptosis of monocyte-derived dendritic cells. Exp. Parasitol. 2016, 163, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-López, R.; Argueta-Donohué, J.; Wilkins-Rodriguez, A.; Escalona-Montaño, A.; Aguirre-García, M.; Gutiérrez-Kobeh, L. Leishmania mexicana amastigotes inhibit p38 and JNK and activate PI3K/AKT: Role in the inhibition of apoptosis of dendritic cells. Parasite Immunol. 2015, 37, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Contreras, I.; Estrada, J.A.; Guak, H.; Martel, C.; Borjian, A.; Ralph, B.; Shio, M.T.; Fournier, S.; Krawczyk, C.M.; Olivier, M. Impact of Leishmania mexicana infection on dendritic cell signalling and functions. PLoS Negl. Trop. Dis. 2014, 8, e3202. [Google Scholar] [CrossRef] [PubMed]
- Prive, C.; Descoteaux, A. Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur. J. Immunol. 2000, 30, 2235–2244. [Google Scholar] [CrossRef]
- Junghae, M.; Raynes, J.G. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect. Immun. 2002, 70, 5026–5035. [Google Scholar] [CrossRef] [PubMed]
- Hallé, M.; Gomez, M.A.; Stuible, M.; Shimizu, H.; McMaster, W.R.; Olivier, M.; Tremblay, M.L. The Leishmania surface protease GP63 cleaves multiple intracellular proteins and actively participates in p38 mitogen-activated protein kinase inactivation. J. Biol. Chem. 2008, 284, 6893–6908. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mosser, D.M.; Zhang, X. Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J. Immunol. 2007, 178, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.J.; Goodridge, H.S.; Harnett, M.M.; Wei, X.Q.; Nikolaev, A.V.; Higson, A.P.; Liew, F.Y. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J. Immunol. 1999, 163, 6403–6412. [Google Scholar] [PubMed]
- Boggiatto, P.M.; Martinez, P.A.; Pullikuth, A.; Jones, D.E.; Bellaire, B.; Catling, A.; Petersen, C. Targeted extracellular signal-regulated kinase activation mediated by Leishmania amazonensis requires MP1 scaffold. Microbes Infect. 2014, 16, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, A.; Leal, N.; Kima, P.E. Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell. Microbiol. 2007, 9, 84–96. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Vieira, T.; Guimarães-Costa, A.; Rochael, N.C.; Lira, M.N.; Nascimento, M.T.; Lima-Gomez, P.D.S.; Mariante, R.M.; Persechini, P.M.; Saraiva, E.M. Neutrophil extracellular traps release induced by Leishmania: Role of PI3K, ERK, PI3K, PKC, and [Ca2+]. J. Leukoc. Biol. 2016, 36, 13–31. [Google Scholar]
- Hong, S.; Lee, H.J.; Kim, S.J.; Hahm, K.B. Connection between inflammation and carcinogenesis in gastrointestinal tract: Focus on TGF-beta signalling. World J. Gastroenterol. 2010, 16, 2080–2093. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signalling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Leof, E.B. TGF-β signalling: A tale of two responses. J. Cell. Biochem. 2007, 102, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Gouman, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell. Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-β signalling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Corneliu, S.C.; Reiss, M.; Danielpour, D. Insulin-like growth factor-I inhibits transcriptional responses of transforming growth factor-β by phosphatidylinositol 3-kinase/Akt-dependent suppression of the activation of Smad3 but not Smad2. J. Biol. Chem. 2003, 278, 38342–38351. [Google Scholar] [CrossRef] [PubMed]
- Loomans, H.A.; Andl, C.D. Intertwining of Activin A and TGFβ Signalling: Dual Roles in Cancer Progression and Cancer Cell Invasion. Cancers 2014, 7, 70–91. [Google Scholar] [CrossRef] [PubMed]
- García, M.J.; Gómez-Reino, C.J. The physiopathology of cyclooxygenase-1 and cyclooxygenase-2. Rev. Esp. Reumatol. 2000, 27, 33–35. [Google Scholar]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ferrández, A.; Prescott, S.; Burt, R.W. COX-2 and colorectal cancer. Curr. Pharm. Des. 2003, 9, 2229–2251. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Fulford, M.; Li, C.F.; Boonyaratanakornkit, J.; Sayyah, S. Arachidonic acid activates phosphatidylinositol 3-kinase signalling and induces gene expression in prostate cancer. Cancer Res. 2006, 66, 1427–1433. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solano-Gálvez, S.G.; Abadi-Chiriti, J.; Gutiérrez-Velez, L.; Rodríguez-Puente, E.; Konstat-Korzenny, E.; Álvarez-Hernández, D.-A.; Franyuti-Kelly, G.; Gutiérrez-Kobeh, L.; Vázquez-López, R. Apoptosis: Activation and Inhibition in Health and Disease. Med. Sci. 2018, 6, 54. https://doi.org/10.3390/medsci6030054
Solano-Gálvez SG, Abadi-Chiriti J, Gutiérrez-Velez L, Rodríguez-Puente E, Konstat-Korzenny E, Álvarez-Hernández D-A, Franyuti-Kelly G, Gutiérrez-Kobeh L, Vázquez-López R. Apoptosis: Activation and Inhibition in Health and Disease. Medical Sciences. 2018; 6(3):54. https://doi.org/10.3390/medsci6030054
Chicago/Turabian StyleSolano-Gálvez, Sandra Georgina, Jack Abadi-Chiriti, Luis Gutiérrez-Velez, Eduardo Rodríguez-Puente, Enrique Konstat-Korzenny, Diego-Abelardo Álvarez-Hernández, Giorgio Franyuti-Kelly, Laila Gutiérrez-Kobeh, and Rosalino Vázquez-López. 2018. "Apoptosis: Activation and Inhibition in Health and Disease" Medical Sciences 6, no. 3: 54. https://doi.org/10.3390/medsci6030054
APA StyleSolano-Gálvez, S. G., Abadi-Chiriti, J., Gutiérrez-Velez, L., Rodríguez-Puente, E., Konstat-Korzenny, E., Álvarez-Hernández, D.-A., Franyuti-Kelly, G., Gutiérrez-Kobeh, L., & Vázquez-López, R. (2018). Apoptosis: Activation and Inhibition in Health and Disease. Medical Sciences, 6(3), 54. https://doi.org/10.3390/medsci6030054