Myc, Oncogenic Protein Translation, and the Role of Polyamines


{kind=link}
Abstract
1. Introduction
2. MYC-Driven Oncogenesis
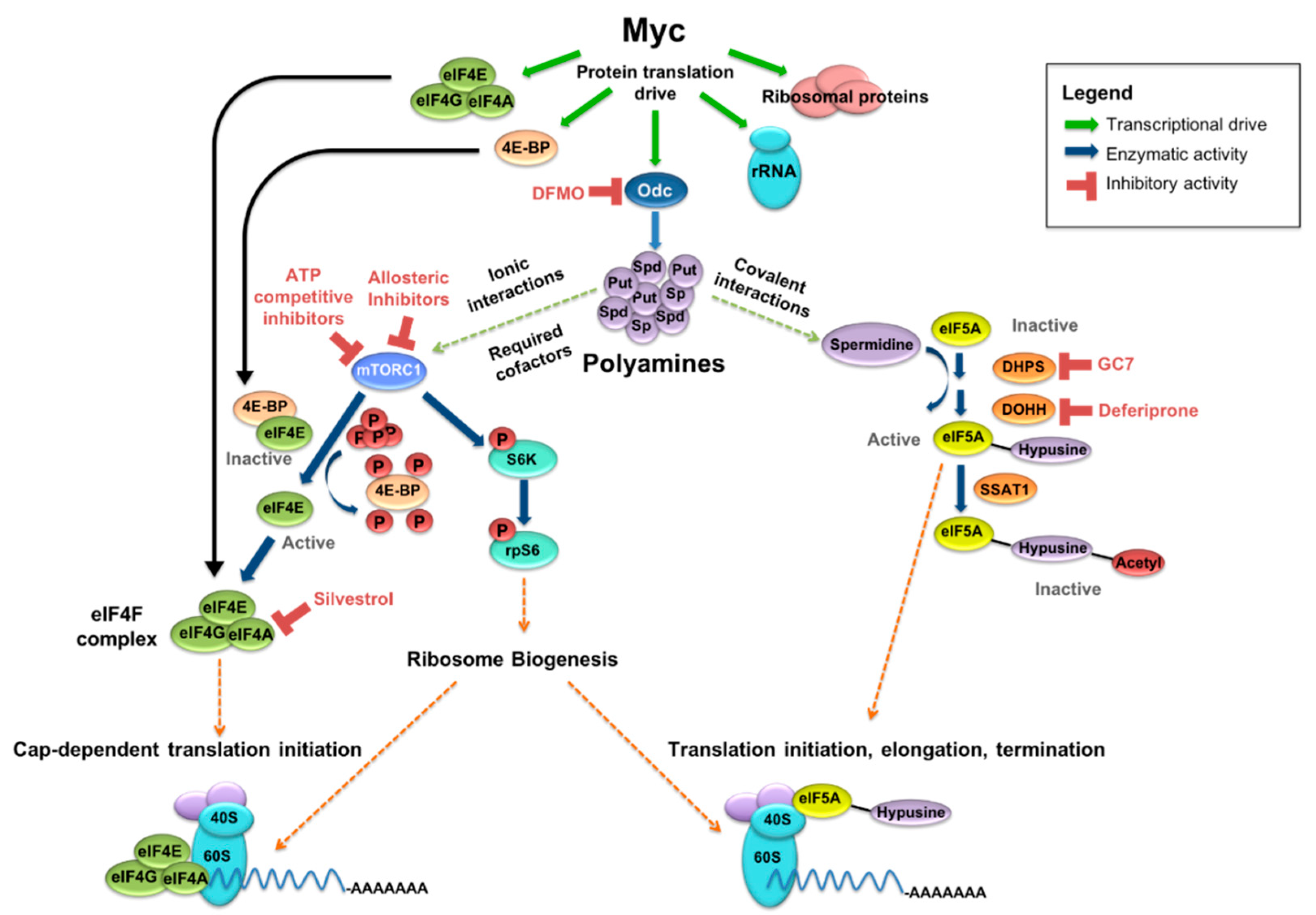
MYC and Protein Synthesis
3. Polyamines
3.1. Polyamine Biosynthesis and Metabolism
3.2. MYC, Polyamines, and Cancer
3.3. Polyamines and Protein Translation
4. Targeting Polyamine Homeostasis and Protein Translation as a Therapy for MYC-Driven Cancers
4.1. Targeting Polyamine Homeostasis
4.2. Targeting Polyamine Uptake and Export
4.3. The eIF4F Complex and Cap-Dependent Translation
4.4. Targeting The eIF4F Complex
4.5. Eukaryotic Translation Initiation Factor 5A and Its Spermidine-Dependent Activation
4.6. Inhibition of eIF5A Hypusination
5. Conclusions: Current Gaps and Future Perspective
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Gerner, E.W.; Meyskens, F.L., Jr.; Goldschmid, S.; Lance, P.; Pelot, D. Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids 2007, 33, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Keller, U.B.; Baudino, T.A.; Yang, C.; Norton, S.; Old, J.A.; Nilsson, L.M.; Neale, G.; Kramer, D.L.; Porter, C.W.; et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell 2005, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Thomas, T.J. Estradiol control of ornithine decarboxylase mRNA, enzyme activity, and polyamine levels in MCF-7 breast cancer cells: Therapeutic implications. Breast Cancer Res. Treat. 1994, 29, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Sheiness, D.K.; Hughes, S.H.; Varmus, H.E.; Stubblefield, E.; Bishop, J.M. The vertebrate homolog of the putative transforming gene of avian myelocytomatosis virus: Characteristics of the DNA locus and its RNA transcript. Virology 1980, 105, 415–424. [Google Scholar] [CrossRef]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Barrans, S.; Crouch, S.; Smith, A.; Turner, K.; Owen, R.; Patmore, R.; Roman, E.; Jack, A. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.; Seeger, R.; Schwab, M.; Varmus, H.; Bishop, J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, A.; Medeiros, L.J.; Abruzzo, L.V.; Lin, P. Lymphoid neoplasms associated with concurrent t(14;18) and 8q24/c-MYC translocation generally have a poor prognosis. Mod. Pathol. 2006, 19, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V.; Vogt, P.K. Therapeutic targeting of Myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Staller, P.; Eilers, M. Control of cell proliferation by Myc. Trends Cell Biol. 1998, 8, 202–206. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Obaya, A.J.; Adachi, S.; Sedivy, J.M. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 1039–1048. [Google Scholar]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-Myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef]
- Pelengaris, S.; Littlewood, T.; Khan, M.; Elia, G.; Evan, G. Reversible activation of c-Myc in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 1999, 3, 565–577. [Google Scholar] [CrossRef]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Mourant, J.R.; Short, K.W.; Carpenter, S.; Kunapareddy, N.; Coburn, L.; Powers, T.M.; Freyer, J.P. Biochemical differences in tumorigenic and nontumorigenic cells measured by Raman and infrared spectroscopy. J. Biomed. Opt. 2005, 10, 031106. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wu, G.; Zhan, X.; Nolan, A.; Koh, C.; De Marzo, A.; Doan, H.M.; Fan, J.; Cheadle, C.; Fallahi, M.; et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011, 6, e26057. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voute, P.A.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.R.; Song, Y.K.; Yu, L.R.; Wei, J.S.; Chung, J.Y.; Hewitt, S.M.; Veenstra, T.D.; Khan, J. Global genomic and proteomic analysis identifies biological pathways related to high-risk neuroblastoma. J. Proteome Res. 2010, 9, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Sahoo, D.; Arvanitis, C.; Bradon, N.; Dill, D.L.; Felsher, D.W. Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet. 2008, 4, e1000090. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Pyronnet, S.; Sonenberg, N. Cell-cycle-dependent translational control. Curr. Opin. Genet. Dev. 2001, 11, 13–18. [Google Scholar] [CrossRef]
- Lin, C.J.; Malina, A.; Pelletier, J. c-Myc and eIF4F constitute a feedforward loop that regulates cell growth: Implications for anticancer therapy. Cancer Res. 2009, 69, 7491–7494. [Google Scholar] [CrossRef] [PubMed]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Functions of polyamines in mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [PubMed]
- Albeck, S.; Dym, O.; Unger, T.; Snapir, Z.; Bercovich, Z.; Kahana, C. Crystallographic and biochemical studies revealing the structural basis for antizyme inhibitor function. Protein Sci. Publ. Protein Soc. 2008, 17, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Kurian, L.; Palanimurugan, R.; Godderz, D.; Dohmen, R.J. Polyamine sensing by nascent ornithine decarboxylase antizyme stimulates decoding of its mRNA. Nature 2011, 477, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Nowotarski, S.L.; Shantz, L.M. Cytoplasmic accumulation of the RNA-binding protein HuR stabilizes the ornithine decarboxylase transcript in a murine nonmelanoma skin cancer model. J. Biol. Chem. 2010, 285, 31885–31894. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Regulation of ornithine decarboxylase. J. Biol. Chem. 2006, 281, 14529–14532. [Google Scholar] [CrossRef] [PubMed]
- Shantz, L.M. Transcriptional and translational control of ornithine decarboxylase during ras transformation. Biochem. J. 2004, 377, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Palanimurugan, R.; Scheel, H.; Hofmann, K.; Dohmen, R.J. Polyamines regulate their synthesis by inducing expression and blocking degradation of ODC antizyme. EMBO J. 2004, 23, 4857–4867. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.L.; Diegelman, P.; Jell, J.; Vujcic, S.; Merali, S.; Porter, C.W. Polyamine acetylation modulates polyamine metabolic flux, a prelude to broader metabolic consequences. J. Biol. Chem. 2008, 283, 4241–4251. [Google Scholar] [CrossRef] [PubMed]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining mysteries of molecular biology: The role of polyamines in the cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef] [PubMed]
- Evageliou, N.F.; Hogarty, M.D. Disrupting polyamine homeostasis as a therapeutic strategy for neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5956–5961. [Google Scholar] [CrossRef] [PubMed]
- Gamble, L.D.; Hogarty, M.D.; Liu, X.; Ziegler, D.S.; Marshall, G.; Norris, M.D.; Haber, M. Polyamine pathway inhibition as a novel therapeutic approach to treating neuroblastoma. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Thomas, T.J. Polyamines in cell growth and cell death: Molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001, 58, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. New Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Fredlund, E.; Ringner, M.; Maris, J.M.; Pahlman, S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 14094–14099. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, P.; Boeva, V.; Hocking, T.D.; Cannoodt, R.; Ambros, I.M.; Ambros, P.F.; Asgharzadeh, S.; Attiyeh, E.F.; Combaret, V.; Defferrari, R.; et al. Genomic amplifications and distal 6q loss: Novel markers for poor survival in high-risk neuroblastoma patients. J. Natl. Cancer Inst. 2018, 110, 1–10. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Kenyon, R.; McGuckin, A.G.; Kohl, N.; Kogner, P.; Christiansen, H.; Pearson, A.D.; Lunec, J. Analysis of candidate gene co-amplification with MYCN in neuroblastoma. Eur. J. Cancer 1997, 33, 2037–2042. [Google Scholar] [CrossRef]
- Auvinen, M.; Paasinen, A.; Andersson, L.C.; Holtta, E. Ornithine decarboxylase activity is critical for cell transformation. Nature 1992, 360, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef] [PubMed]
- Moshier, J.A.; Dosescu, J.; Skunca, M.; Luk, G.D. Transformation of NIH/3T3 cells by ornithine decarboxylase overexpression. Cancer Res. 1993, 53, 2618–2622. [Google Scholar] [PubMed]
- Landau, G.; Bercovich, Z.; Park, M.H.; Kahana, C. The role of polyamines in supporting growth of mammalian cells is mediated through their requirement for translation initiation and elongation. J. Biol. Chem. 2010, 285, 12474–12481. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.F.; Lewis, J.B.; Anderson, C.W.; Gesteland, R.F. Enhanced differential synthesis of proteins in a mammalian cell-free system by addition of polyamines. J. Biol. Chem. 1975, 250, 5688–5695. [Google Scholar] [PubMed]
- Igarashi, K.; Hikami, K.; Sugawara, K.; Hirose, S. Effect of polyamines on polypeptide synthesis in rat liver cell-free system. Biochim. Biophys. Acta 1973, 299, 325–330. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of protein synthesis by polyamines. IUBMB Life 2015, 67, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Mandal, A.; Johansson, H.E.; Orjalo, A.V.; Park, M.H. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Huynh, D.; Shen, Y.J.; Zhou, J.; Yusufzai, T.; Salem, K.Z.; Ebright, R.Y.; Shi, J.; Park, J.; Glavey, S.V.; et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Evageliou, N.F.; Haber, M.; Vu, A.; Laetsch, T.W.; Murray, J.; Gamble, L.D.; Cheng, N.C.; Liu, K.; Reese, M.; Corrigan, K.A.; et al. Polyamine antagonist therapies inhibit neuroblastoma initiation and progression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4391–4404. [Google Scholar] [CrossRef] [PubMed]
- Rounbehler, R.J.; Li, W.; Hall, M.A.; Yang, C.; Fallahi, M.; Cleveland, J.L. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 2009, 69, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martin-Martin, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Samal, K.; Zhao, P.; Kendzicky, A.; Yco, L.P.; McClung, H.; Gerner, E.; Burns, M.; Bachmann, A.S.; Sholler, G. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int. J. Cancer 2013, 133, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Gitto, S.B.; Pandey, V.; Oyer, J.L.; Copik, A.J.; Hogan, F.C.; Phanstiel, O.; Altomare, D.A. Difluoromethylornithine combined with a polyamine transport inhibitor is effective against gemcitabine resistant pancreatic cancer. Mol. Pharm. 2018, 15, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Malka-Mahieu, H.; Newman, M.; Desaubry, L.; Robert, C.; Vagner, S. Molecular pathways: The eIF4F translation initiation complex-new opportunities for cancer treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Lazaris-Karatzas, A.; Sonenberg, N. The mRNA 5′ cap-binding protein, eIF-4E, cooperates with v-myc or E1A in the transformation of primary rodent fibroblasts. Mol. Cell. Biol. 1992, 12, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D.; Montanaro, L.; Ma, L.; Xu, W.; Londei, P.; Cordon-Cardo, C.; Pandolfi, P.P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med. 2004, 10, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Lazaris-Karatzas, A.; Sonenberg, N. Mrnas containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992, 11, 4153–4158. [Google Scholar] [PubMed]
- Larsson, O.; Li, S.; Issaenko, O.A.; Avdulov, S.; Peterson, M.; Smith, K.; Bitterman, P.B.; Polunovsky, V.A. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007, 67, 6814–6824. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Jaramillo, M.; Liu, Y.L.; Dever, T.E.; Merrick, W.C.; Kung, H.F.; Sonenberg, N. Translation initiation factors induce DNA synthesis and transform NIH 3T3 cells. New Biol. 1990, 2, 648–654. [Google Scholar] [PubMed]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Shantz, L.M.; Coleman, C.S.; Pegg, A.E. Expression of an ornithine decarboxylase dominant-negative mutant reverses eukaryotic initiation factor 4E-induced cell transformation. Cancer Res. 1996, 56, 5136–5140. [Google Scholar] [PubMed]
- Lin, C.J.; Cencic, R.; Mills, J.R.; Robert, F.; Pelletier, J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008, 68, 5326–5334. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, X.; Yin, Y.; Li, X.; Gao, H.; Bazer, F.W.; Wu, G. Putrescine stimulates the mTOR signaling pathway and protein synthesis in porcine trophectoderm cells. Biol. Reprod. 2014, 91, 106. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Shimogori, T.; Suzuki, T.; Kashiwagi, K.; Kakinuma, Y.; Igarashi, K. Enhancement of helicase activity and increase of eIF-4E phosphorylation in ornithine decarboxylase-overproducing cells. Biochem. Biophys. Res. Commun. 1996, 222, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Oh, Y.; Wheler, J.; Naing, A.; Brail, L.; Callies, S.; Andre, V.; Kadam, S.K.; Nasir, A.; et al. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6582–6591. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, G.; Xu, Q.; Rudolph-Owen, L.; Tendyke, K.; Liu, J.; Towle, M.; Zhao, N.; Marsh, J.; Agoulnik, S.; Twine, N.; et al. Potent in vitro and in vivo anticancer activities of des-methyl, des-amino pateamine A, a synthetic analogue of marine natural product pateamine A. Mol. Cancer Ther. 2009, 8, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Cencic, R.; Robert, F.; Galicia-Vazquez, G.; Malina, A.; Ravindar, K.; Somaiah, R.; Pierre, P.; Tanaka, J.; Deslongchamps, P.; Pelletier, J. Modifying chemotherapy response by targeted inhibition of eukaryotic initiation factor 4A. Blood Cancer J. 2013, 3, e128. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.E.; Robert, F.; Gerard, B.; Lindqvist, L.; Chen, S.M.; Wendel, H.G.; Brem, B.; Greger, H.; Lowe, S.W.; Porco, J.A.; et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Investig. 2008, 118, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, Z.A.; Haag, P.G.; Johansson, H.E. Human eIF5A2 on chromosome 3q25-q27 is a phylogenetically conserved vertebrate variant of eukaryotic translation initiation factor 5A with tissue-specific expression. Genomics 2001, 71, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Benne, R.; Hershey, J.W. The mechanism of action of protein synthesis initiation factors from rabbit reticulocytes. J. Biol. Chem. 1978, 253, 3078–3087. [Google Scholar] [PubMed]
- Schreier, M.H.; Erni, B.; Staehelin, T. Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors. J. Mol. Biol. 1977, 116, 727–753. [Google Scholar] [CrossRef]
- Henderson, A.; Hershey, J.W. Eukaryotic translation initiation factor (eIF) 5A stimulates protein synthesis in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 6415–6419. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Eyler, D.E.; Green, R.; Dever, T.E. Hypusine-containing protein eIF5A promotes translation elongation. Nature 2009, 459, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Doerfel, L.K.; Wohlgemuth, I.; Kothe, C.; Peske, F.; Urlaub, H.; Rodnina, M.V. Ef-p is essential for rapid synthesis of proteins containing consecutive proline residues. Science 2013, 339, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Ude, S.; Lassak, J.; Starosta, A.L.; Kraxenberger, T.; Wilson, D.N.; Jung, K. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 2013, 339, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.; Shin, B.S.; Woolstenhulme, C.J.; Kim, J.R.; Saini, P.; Buskirk, A.R.; Dever, T.E. eIF5A promotes translation of polyproline motifs. Mol. Cell 2013, 51, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.P.; Wu, C.C.; Dever, T.E.; Buskirk, A.R.; Green, R. eIF5A functions globally in translation elongation and termination. Mol. Cell 2017, 66, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Mandal, S.; Park, M.H. Genome-wide analyses and functional classification of proline repeat-rich proteins: Potential role of eIF5A in eukaryotic evolution. PLoS ONE 2014, 9, e111800. [Google Scholar] [CrossRef] [PubMed]
- Wolff, E.C.; Kang, K.R.; Kim, Y.S.; Park, M.H. Posttranslational synthesis of hypusine: Evolutionary progression and specificity of the hypusine modification. Amino Acids 2007, 33, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Pallmann, N.; Braig, M.; Sievert, H.; Preukschas, M.; Hermans-Borgmeyer, I.; Schweizer, M.; Nagel, C.H.; Neumann, M.; Wild, P.; Haralambieva, E.; et al. Biological relevance and therapeutic potential of the hypusine modification system. J. Biol. Chem. 2015, 290, 18343–18360. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.L.; Park, M.H.; Folk, J.E.; Safer, B.; Braverman, R. Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc. Natl. Acad. Sci. USA 1983, 80, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Park, J.H.; Folk, J.E.; Deck, J.A.; Pegg, A.E.; Sokabe, M.; Fraser, C.S.; Park, M.H. Inactivation of eukaryotic initiation factor 5A (eIF5A) by specific acetylation of its hypusine residue by spermidine/spermine acetyltransferase 1 (SSAT1). Biochem. J. 2011, 433, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Cleveland, J.L. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids 2016, 48, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Scuoppo, C.; Miething, C.; Lindqvist, L.; Reyes, J.; Ruse, C.; Appelmann, I.; Yoon, S.; Krasnitz, A.; Teruya-Feldstein, J.; Pappin, D.; et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature 2012, 487, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, S.; Gontarewicz, A.; Ziegler, P.; Hartmann, U.; Kammer, W.; Copland, M.; Brassat, U.; Priemer, M.; Hauber, I.; Wilhelm, T.; et al. Hypusination of eukaryotic initiation factor 5A (eIF5A): A novel therapeutic target in BCR-ABL-positive leukemias identified by a proteomics approach. Blood 2007, 109, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Preukschas, M.; Hagel, C.; Schulte, A.; Weber, K.; Lamszus, K.; Sievert, H.; Pallmann, N.; Bokemeyer, C.; Hauber, J.; Braig, M.; et al. Expression of eukaryotic initiation factor 5A and hypusine forming enzymes in glioblastoma patient samples: Implications for new targeted therapies. PLoS ONE 2012, 7, e43468. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Yu, H.; Shi, X.; Sun, L.; Zhou, Q.; Zheng, D.; Shi, H.; Li, N.; Zhang, X.; Shao, G. Cisplatin sensitivity is enhanced in non-small cell lung cancer cells by regulating epithelial-mesenchymal transition through inhibition of eukaryotic translation initiation factor 5A2. BMC Pulm. Med. 2014, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shen, T.; Luo, Y.; Liu, L.; Chen, W.; Xu, B.; Han, X.; Pang, J.; Rivera, C.A.; Huang, S. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer 2010, 127, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Schmidt, M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G. Targeting the wnt/beta-catenin pathway with the antifungal agent ciclopirox olamine in a murine myeloma model. In Vivo 2011, 25, 887–893. [Google Scholar] [PubMed]
- Memin, E.; Hoque, M.; Jain, M.R.; Heller, D.S.; Li, H.; Cracchiolo, B.; Hanauske-Abel, H.M.; Pe’ery, T.; Mathews, M.B. Blocking eIF5A modification in cervical cancer cells alters the expression of cancer-related genes and suppresses cell proliferation. Cancer Res. 2014, 74, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Minden, M.D.; Hogge, D.E.; Weir, S.J.; Kasper, J.; Webster, D.A.; Patton, L.; Jitkova, Y.; Hurren, R.; Gronda, M.; Goard, C.A.; et al. Oral ciclopirox olamine displays biological activity in a phase i study in patients with advanced hematologic malignancies. Am. J. Hematol. 2014, 89, 363–368. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flynn, A.T.; Hogarty, M.D. Myc, Oncogenic Protein Translation, and the Role of Polyamines. Med. Sci. 2018, 6, 41. https://doi.org/10.3390/medsci6020041
Flynn AT, Hogarty MD. Myc, Oncogenic Protein Translation, and the Role of Polyamines. Medical Sciences. 2018; 6(2):41. https://doi.org/10.3390/medsci6020041
Chicago/Turabian StyleFlynn, Andrea T., and Michael D. Hogarty. 2018. "Myc, Oncogenic Protein Translation, and the Role of Polyamines" Medical Sciences 6, no. 2: 41. https://doi.org/10.3390/medsci6020041
APA StyleFlynn, A. T., & Hogarty, M. D. (2018). Myc, Oncogenic Protein Translation, and the Role of Polyamines. Medical Sciences, 6(2), 41. https://doi.org/10.3390/medsci6020041