Abstract
Multiple sclerosis (MS) is one of the most common neurological disorders in young adults. The etiology of MS is not known but it is widely accepted that it is autoimmune in nature. Disease onset is believed to be initiated by the activation of CD4+ T cells that target autoantigens of the central nervous system (CNS) and their infiltration into the CNS, followed by the expansion of local and infiltrated peripheral effector myeloid cells that create an inflammatory milieu within the CNS, which ultimately lead to tissue damage and demyelination. Clinical studies have shown that progression of MS correlates with the abnormal expression of certain cytokines. The use of experimental autoimmune encephalomyelitis (EAE) model further delineates the role of these cytokines in neuroinflammation and the therapeutic potential of manipulating their biological activity in vivo. In this review, we will first present an overview on cytokines that may contribute to the pathogenesis of MS or EAE, and provide successful examples and roadblock of translating data obtained from EAE to MS. We will then focus in depth on recent findings that demonstrate the pathological role of granulocyte-macrophage colony-stimulating factor (GM-CSF) in MS and EAE, and briefly discuss the potential of targeting effector myeloid cells as a treatment strategy for MS.
1. Introduction
Multiple sclerosis (MS) is one of the most common neurological diseases in young adults, with initial clinical signs often observed between 20 and 45 years of age [1]. It is more prevalent in females than in males, with incidence ratio of about 3:1 [2]. The exact mechanism accounting for this gender difference is unclear, but possibly due to the effect of gonadal hormones. Currently, more than two million people suffer from MS worldwide and over 400,000 people are affected in the United States [3,4]. Given the early onset and lifelong progression of the disease, MS poses significant psychological burden to the patients and their families, as well as economic burden to society [5]. Characteristics of MS pathology include plaque formation in the central nervous system (CNS) due to inflammation and demyelination, which result in clinical manifestations such as vision impairment, cognitive problems, loss of muscle coordination, weakness and fatigue, numbness, depression, bowel changes, and bladder dysfunction, depending on which area(s) of the CNS is affected [1,6,7].
2. Diagnosis and Clinical Courses of Multiple Sclerosis
The wide range of clinical manifestations of MS overlaps with other types of neurological disorders, such as acute disseminated encephalomyelitis and neuromyelitis optica spectrum disorder. Therefore, early and precise diagnosis is needed for proper disease management and treatments. Diagnosis of MS is heavily based on results from magnetic resonance imaging (MRI) depicting lesion(s) dissemination in space (DIS) and time (DIT) [8]. MRI is also a valuable tool for monitoring disease progression and effectiveness of treatments, based on the number of newly formed lesions [9]. In addition, paraclinical tests showing evidence of elevated immunoglobulin G (IgG) index or evidence of oligoclonal bands in the cerebrospinal fluid (CSF) further confirms certain cases of MS [10].
The clinical course of MS varies among patients and is categorized based on the frequency of attacks (relapses) and the patterns of disease progression [11]. Relapsing-remitting MS (RRMS) is the most prevalent subtype which affects ~85% of patients [1]. The clinical pattern of RRMS is marked by unpredictable relapses resulting in acute neurological disability, followed by disease remission. Patients may have partial or complete recovery during each remission [1,12]. Unfortunately, disease pattern changes over time and patients suffering from RRMS eventually develop secondary progressive MS (SPMS), characterized by slow but significant increase in disease severity without remission [13]. Primary progressive MS (PPMS) affects ~10% of MS patients, which is characterized by progressive deterioration of neurological functions from the onset of disease symptoms without relapse and remission [14,15]. Pathological and MRI features of SPMS and PPMS are indistinguishable, suggesting the lack of relapsing-remitting phase in PPMS may be due to attacks at the clinically silent regions during the early stage of the disease. Once damage accumulates in the CNS, clinical manifestations of MS become apparent [16,17].
3. Multiple Sclerosis as an Immune-Mediated Disease
The etiology of MS is not well-defined but likely multifactorial, involving both genetic and environmental factors. MS is generally considered an autoimmune disease, with support from several lines of evidence. Genome-wide association studies (GWAS) identified over 100 MS risk loci, many of which overlap with those identified from other autoimmune diseases [18,19]. The majority of these loci regulate or encode genes that control immune cell functions, such as human leukocyte antigen (HLA) [20]. Moreover, immune cells infiltrating from the periphery, especially T cells and myeloid cells, are often identified around the demyelinating plaques [21,22]. Importantly, although the frequency of peripheral myelin-reactive T cells is comparable between MS patients and healthy donors, those cells from the patients express higher amounts of pro-inflammatory cytokines [23], supporting the autoimmune nature of MS.
Vitamin D deficiency and Epstein-Barr virus (EBV) infection are considered the most relevant extrinsic risk factors of MS, and both of them to some extent are due to their dysregulating effects on the immune system. The bioactive form of vitamin D, i.e., 1, 25 dihydroxyvitamin D, regulates both innate and adaptive immune responses by modulating T helper (Th)-cell and B-cell differentiation, while also maintaining the tolerogenic status of dendritic cells (DCs), which suppress T-cell activation [24,25,26]. In addition, recent studies suggest that polymorphisms of the vitamin D receptor gene are associated with the incidence of MS [27,28]. MS patients often test positive in serological analysis for EBV [29,30]. EBV is a human herpesvirus capable of infecting B cells, resulting in infectious mononucleosis in adults [31]. It is not clear how EBV infection links to the pathogenesis of MS, though there is speculation that infection mediates molecular mimicry and/or bystander activation of antigen-presenting cells (APCs) and autoreactive T cells [32].
The most convincing evidence to support the immune-mediated nature of MS comes from the fact that the two most effective disease-modifying drugs currently available for RRMS, natalizumab and fingolimod, block leukocyte egress from the periphery and infiltration into the CNS [33]. Natalizumab blocks the interaction of very late activation antigen (VLA)-4 expressed on Th1 cells with the vascular cell adhesion molecule (VCAM)-1, thus preventing extravasation of these cells [34]. Fingolimod, on the other hand, down-regulates sphingosine-1-phosphate receptor 1 (S1PR1) and selectively traps the CCR7+ central memory T cells in the lymph nodes [35]. The relatively high efficacy (55–65% reduction in relapses) of these drugs also suggests that MS is initiated by abnormal activation of peripheral T cells [36,37].
4. Experimental Autoimmune Encephalomyelitis Is a Valuable Model for Multiple Sclerosis Research
Research over the past few decades has focused on identifying immune cell types and mediators involved in the pathogenesis of MS, with an ultimate goal of improving clinical outcomes. Although scientists and clinicians have learned much by analyzing the cellular and molecular profiles of CSF and peripheral blood from the patients, as well as postmortem tissue examination, the causal relationship between experimental results and disease initiation and progression is difficult to establish. Clinical trials of mechanistically well-defined drugs are one way to identify critical components contributing to the pathogenesis of MS, but these trials are often difficult to initiate without support from experimental data. Therefore, animal models play a critical role in not only dissecting the molecular pathways governing the pathogenesis of MS, but also for identifying new therapeutic agents. Experimental autoimmune encephalomyelitis (EAE) is the best animal model available to study MS [38,39]. EAE can be induced in various species but mice are most often used given the vast repertoire of genetically-modified animals [39]. Neuroinflammation, subsequent paralysis and physical disability can be induced by active immunization of mice with self-antigen derived from the CNS, including myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), or proteolipid protein (PLP), emulsified in complete Freund’s adjuvant (CFA) [39]. Further studies showed that immunization with antigenic epitopes of those proteins, such as MOG35–55 and PLP139–151 peptides, results in EAE with comparable disease severity [40,41,42]. Active immunization is often supplemented with injections of pertussis toxin, which facilitate the recruitment of leukocytes to the CNS and increase permeability of the blood–brain barrier (BBB) [43]. Recently, it was reported that injection of pertussis toxin drives an early production of interleukin (IL)-1β, which is important for the priming phase of EAE [44]. Depending on the mouse strains, in addition to the choice and dosages of peptides, patterns of EAE can be manipulated from relapsing-remitting to progressive subtypes, resembling MS pathology in humans [39]. EAE can also be induced by adoptive transfer of the autoreactive CD4+ T cells purified from actively immunized mice, or CD4+ T cells isolated from the transgenic mouse strain in which the T-cell receptor (TCR) specifically recognizes MOG35–55 (the 2D2 mice) [45,46]. In the latter case, CD4+ T cells are purified from the unimmunized mice, and then skewed to different T-helper lineages in vitro before injecting these cells to the recipient mice [39]. This approach is particularly useful to dissect the distinctive roles of Th1 and Th17 cells in neuroinflammation. Indeed, EAE was one of the first disease models used to elucidate the pathogenic role of Th17 cells [47,48].
Besides the elucidation of immune cell types and molecular pathways governing the pathogenesis of neuroinflammation, EAE model has also been widely used for testing potential therapeutic agents for MS, and some of them have been successfully translated to the clinic. Glatiramer acetate (GA, Copaxone, Teva Pharmaceutical Industries Ltd., Petah Tikva, Israel), a first-generation MS drug, was first proven to effectively suppress EAE [49,50]. GA is a random copolymer that is comprised of four amino acids found in MBP: tyrosine, glutamate, alanine, and lysine [51]. It was predicted that GA would be encephalitogenic given its sequence similarity with MBP, but it is surprisingly immunosuppressive [49,50]. After over 20 years of animal research, a phase III multicenter clinical trial demonstrated that subcutaneous injections of GA reduce relapse rate by 29% in patients with RRMS and improves neurologic disability [52,53]. Exactly how GA improves EAE and MS outcomes is not clear but EAE studies suggest that multiple mechanisms are involved, including suppression of Th1 and Th17 cell-mediated inflammation; induction of regulatory T cell (Treg) differentiation [54,55,56,57] and the suppressor function of the CD11b+ Ly6G− monocytes [58]; increasing proliferation, differentiation and survival of oligodendrocyte progenitor cells [59]; and enhancing production of neuroprotection and regeneration factors in the brain [60].
Natalizumab is another MS drug that was discovered through EAE model [61]. Natalizumab is a second-generation drug for RRMS targeting α4-integrin expressed on CD4+ T cells, which together with the β1 subunit, form VLA-4. VLA-4 is important for CD4+ T cells entering the brain parenchyma from the blood during the initiation of neuroinflammation [62]. Clinical trials showed that natalizumab treatments lead to fewer brain lesions and relapses in patients with MS [63], and reduce the risk of sustained progression of disability to about 40% over two years [64]. Unlike GA, natalizumab is a mechanistically well-defined drug discovered through molecular targeting using EAE model, further emphasizing the importance of this model in translational MS research.
Similar to other disease models, EAE model has its limitations and does not perfectly mirror MS in humans. EAE is induced by active immunization of CNS antigens with CFA or by adoptive transfer of encephalitogenic CD4+ T cells to laboratory mice with identical genetic background [65]. MS, on the other hand, is strongly influenced by genetic polymorphisms of individuals and is triggered by ill-defined environmental factors [38]. Therefore, EAE is not a good model for understanding the initiation and prevention of MS. Mechanistically, autoreactive CD4+ T cells play a major role in the pathogenesis of EAE, which are predominately found in the CNS lesions [66], and the role of CD8+ T cells and B cells are often shadowed. However, these cells significantly contribute to the MS pathology [67]. In addition, inflammation and lesions are predominately found at the spinal cord after EAE induction, and thus disease severity is mainly evaluated by the physical disability of the mice due to muscle paralysis [68]. MS patients have diverse clinical manifestations which is indicative of lesions at both brain parenchyma and spinal cord. Thus, EAE may not be a suitable model to study cognitive impairments caused by lesions in the brain in MS patients.
One of the major concerns of translating new therapeutics from EAE to MS is that the side effects of some drugs, which may be severe and even fatal in humans, are not observed in animal models. This may be due to a number of reasons, including different dosages being administered to animals and humans, severe side effects not being observed in small scale animal studies, or the absence of environmental factors that trigger the side effect in controlled animal facility. A prominent example indicating the severity of side effect are reports of progressive multifocal leukoencephalopathy (PML) in MS patients treated with natalizumab [69], leading to a temporary withdrawal of this drug from the market [70]. PML is a rare disease caused by oligodendroglial infection with opportunistic John Cunningham (JC) virus, which is present in the microflora of ~50% of human populations [71]. Although natalizumab was later re-introduced to the clinic for treating MS patients who do not carry JC virus [72], this example reminds clinicians and scientists that carefully designed clinical trials and post-market evaluations of new MS therapeutics are necessary even when promising results are obtained from EAE model.
Besides EAE model, viral- and toxin-induced demyelination models are also used for MS studies. Infectious agents are possible triggers of MS, which is supported by reports showing that some viruses can cause CNS encephalomyelitis [73]. Theiler’s murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD) is an example of a viral-induced animal model of MS. TMEV is a single-stranded RNA virus which belongs to cardiovirus genus of Picornaviridae family [74,75]. Based on neurovirulence, TMEV is classified into two subgroups. The high virulence group consists of GDVII and FA strains which can cause acute encephalitis and death of animal within 1 to 2 weeks of injection [76,77]. The less virulent group is Theiler’s original (TO) subgroup which includes BeAn 8386 (BeAn) and Daniels (DA) strains, can cause acute encephalitis after a week of injection and a more chronic phase of disease a month after infection [75,76,78]. Similar to EAE, TMEV-induced demyelination is through an immune-mediated mechanism and the role of cytokines in this model will be also discussed briefly.
Cuprizone (bis-cyclohexanone-oxalyldihydrazone, CPZ) and lysolecithin are the most commonly used toxins for inducing demyelination in the animal models of MS [79,80,81]. Toxin-induced animal models of MS provide a better scope for studying the non-immune-mediated demyelination and remyelination events [82]. In CPZ-induced animal model of demyelination, susceptible strains of mice are fed with 0.2% of cuprizone-supplemented diets for 4 weeks, which results in demyelination in various regions of the brain, mainly due to the death of oligodendrocytes [79,80,83]. Withdrawal of CPZ from the diets results in spontaneous remyelination. However, if cuprizone diets are continued for 12 weeks, it results in induction of chronic demyelinating lesions [79,81]. CPZ model involves non-inflammatory-mediated demyelination by inducing metabolic stress in oligodendrocytes. This leads to apoptosis of these cells and myelin destruction, and ultimately causes axonal and neuronal pathology [80,84]. Local injection of lysolecithin into spinal cord of animal results in induction of focal areas of demyelination [79], which is another non-immune-mediated model for studying the process of demyelination and remyelination [85]. Cytokines contributing to the pathogenesis of the toxin-induced demyelination is less clear.
5. Cytokines Involved in the Pathogenesis of Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
Although the precise mechanism for the initiation of MS is still not clear, the pathogenic cascade leading to EAE has been well studied and reviewed in detail [86]. Briefly, autoreactive CD4+ T cells are differentiated into pathogenic Th1 and Th17 cells in the secondary lymphoid organs, then egress to the CNS via crossing the choroid plexus. These T cells interact with the APCs residing at the subarachnoid and perivascular space, leading to the expansion of autoreactive T cells, production of pro-inflammatory cytokines, increasing subpial blood vessel permeability, and causing influx of circulating autoreactive T cells and effector myeloid cells. The massive production of pro-inflammatory cytokines by the infiltrated immune cells ultimately destroy BBB integrity, resulting in inflammation and tissue damage [38,78]. Recent transcriptome analysis demonstrated that the myelin-specific CCR6+ T cells from MS patients share gene signatures with the EAE-driven pathogenic Th17 cells [23], suggesting that the above paradigm is to some extent applicable to the pathogenesis of MS.
Cytokines are critically involved throughout the course of MS, from the initial pathogenic T-cell differentiation in the periphery, to the resulting inflammation and tissue damage in the CNS (Table 1). Manipulation of cytokine availability and/or signaling is an attractive strategy for MS treatment as this approach is currently used for treating different autoimmune diseases. For example, tumor necrosis factor (TNF)-α inhibitors are used for treating rheumatoid arthritis and Crohn’s disease. Early studies showed that TNF-α level is elevated in the CSF of MS patients, and correlates with both disease severity and progression [87,88]. Moreover, antibody neutralizing TNF-α prevents EAE induced by adoptive transfer of MBP-specific T cell line [89]. Further, increased expression of TNF-α was observed in the CNS of SJL/J mice infected with TMEV [90]. Treatment of TMEV-infected mice with antibody specific for TNF-α at the onset of disease results in suppression of disease development [91]. This suggests that TNF-α inhibitors might also be effective in treating MS. Unfortunately, clinical trials showed that MS patients treated with recombinant TNF receptor lenercept (sTNFR-IgG p55) experienced exacerbated disease pathology [92], indicating TNF-α may have multiple, cell type-specific roles in neuroinflammation, some of which may be neuro-protective [93].

Table 1.
Major cytokines contributing to the pathogenesis of multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE).
Disease amelioration may also be achieved by elevating the level of cytokines that suppress immune functions. Interferon (IFN)-β1b was the first approved drug for treating RRMS in 1993. At that time, the mechanism in which IFN-β reduces relapse rate was unclear, but it was thought to be related to its anti-viral effects [94,95]. Therefore, the effect of IFN-γ in patients with RRMS was also tested but disease exacerbation was observed [96].
Further studies have shown that IFN-β indeed suppresses neuroinflammation via multiple mechanisms, including the induction of Tregs and suppression of Th17 cell differentiation [97,98]; reduction of the inflammatory cell migration into the CNS [99]; suppression of DC activity [100]; and the induction of IL-10 [101]. The ability of type I interferon to induce IL-10 production is particularly of interest as it has been shown that the IL-10-producing regulatory B cells (B10 cells) significantly reduce EAE severity [102]. ATX-MS-1467 is a mixture of four peptides of human MBP, and injection of these peptides inhibits EAE correlated with IL-10 induction [103]. A phase 2a clinical trial of this agent in RRMS was recently completed (NCT01973491) and results are expected to be released soon.
6. The IL-23-IL17 Axis in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
Targeting cytokines controlling CD4+ T-cell differentiation and effector functions is certainly a rational strategy to ameliorate MS. Prior to the emergence of the concept of Th17 lineage, Th1 cells were the main target. However, several studies have showed that IFN-γ—deficient mice have higher susceptibility to EAE [104,105,106], and IFN-γ unpredictably was found to suppress the expansion of CD4+ T cells during the course of EAE [104]. In TMEV model of demyelination, treatment with anti-IFN-γ antibody results in increased disease severity in SJL/J mice [107]. IL-12, composed by the p40 and p35 subunits, is the driver of Th1 differentiation [108,109]. Surprisingly, although mice lacking IL-12p40 are resistant to EAE, mice lacking IL-12p35 are susceptible [110,111], suggesting that the IL-12-Th1 axis may not be a major contributor to the EAE pathology, and IL-12p40 may have a biological function other than dimerizing with IL-12p35. This question was soon resolved by the discovery that the p40 subunit can dimerize with a p19 subunit, and together they form IL-23. Correspondingly, IL-23p19-deficient mice are resistant to EAE. Further studies showed that the IL-23-dependent CD4+ T cells are highly pathogenic and are characterized by the production of IL-17, IL-6, and TNF [112]. Later the same year, the concept of a distinct Th17 lineage was proposed [48,113] and there was intense interest in understanding the role of these cells in autoimmunity. In TMEV model, Th17 cells contribute to the viral persistence and chronic demyelination, and IL-17 neutralization increases virus clearance and enhances cytotoxic T cell functions [114]. Th17 cells are characterized by the production of IL-17A, IL-17F, IL-21, and IL-22, and can be differentiated in vitro by the transforming growth factor (TGF)-β and IL-6 or IL-21 without the need of IL-23 [115,116,117,118]. However, adoptive transfer of Th17 cells differentiated by TGF-β1 and IL-6 are unable to induce severe EAE is the absence of IL-23 [119]. Correspondingly, disease severity was only partially reduced when EAE was induced by active immunization in IL-17—deficient mice, or by adoptive transfer of lymphocytes isolated from Il17a−/− mice [120,121], suggesting there may be an additional function of IL-23 in neuroinflammation onset. This was shown by studies demonstrating that IL-23 induces the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) in IL-17 cells, and GM-CSF is the main pathogenic factor of EAE [120,122].
7. GM-CSF Connects the Priming Autoreactive CD4+ T Cells to the Effector Myeloid Cells
GM-CSF is not a new mediator of MS and EAE. McQualter et al. [123] in 2001 reported that Csf2−/− mice (which encodes GM-CSF) are resistant to EAE development, correlating with reduced immune cell infiltration into to CNS and diminished proliferation of MOG35–55-specific splenic lymphocytes. Intraperitoneal injection of anti-GM-CSF monoclonal antibody (mAb) significantly reduces EAE severity, suggesting it has promising therapeutic potential [123]. In humans, myelin-reactive CD4+ T cells isolated from MS patients demonstrate an increase in the production of GM-CSF [23], along with an increase in the frequency of GM-CSF-producing memory Th cells [124,125,126]. Importantly, MS patients treated with various disease-modifying drugs, including IFN-β, natalizumab, and GA, have reduced frequencies of the GM-CSF-producing T cells [125,126], further supporting the rationale of targeting GM-CSF signaling as MS therapy.
Understanding the signaling pathways triggered by GM-CSF and its biological functions is critical for designing novel therapeutics with minimal side effects. GM-CSF is a monomeric glycoprotein which was first cloned in mice in 1984 and then in humans in 1985 [127,128]. GM-CSF is primarily produced by T cells, macrophages, mast cells, fibroblasts, and endothelial cells [129]. The GM-CSF receptor (GM-CSFR) is composed by the α chain which is GM-CSF-specific, and the common β chain (βc) that is shared with IL-3 and IL-5 [130]. GM-CSFR is expressed in multiple lineages of cells including monocytes, macrophages, neutrophils, DCs, endothelial cells, and others (Figure 1). GM-CSFR is associated with the tyrosine kinase JAK2 [131]. GM-CSF binding leads to autophosphorylation of JAK2, followed by phosphorylation and activation of the downstream signal transducer of activation of transcription (STAT) proteins. STAT5 is the major STAT protein activated by GM-CSF, which drives transcription of genes responsible for immune cell differentiation and the onset of inflammation [132]. GM-CSF also stimulates the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ), mitogen activated protein kinase (MAP kinase), and phosphoinositide 3 kinase (PI3K) pathways [133].
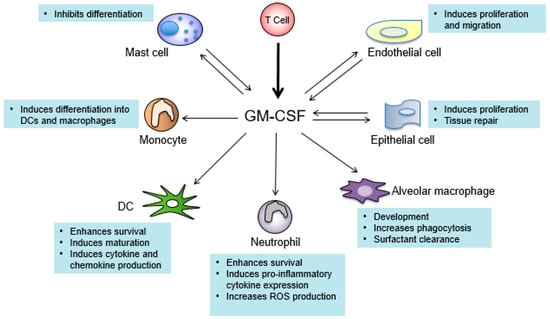
Figure 1.
Cell types that produce and respond to granulocyte-macrophage colony-stimulating factor (GM-CSF). Arrows pointing toward GM-CSF indicate cell types that produce GM-CSF. Arrows pointing away from GM-CSF indicate cells that respond to GM-CSF. T cells (bold arrow) are the main producers of GM-CSF. Texts describe the major functions of GM-CSF in the indicated cell types. DC: dendritic cell; ROS: reactive oxygen species.
Under non-inflammatory condition, GM-CSF is critical for the differentiation of alveolar macrophages (AMs) via the regulation of transcription factor PU.1 [134,135]. Csf2−/− mice showed defects in innate immune functions [135]. Importantly, mutation of GM-CSFRα or the common β chain in humans results in pulmonary alveolar proteinosis (PAP), which is due to defective development of AMs and reduction of surfactant clearance [136,137]. In addition, GM-CSF neutralizing antibodies have been found in the bronchoalveolar lavage fluid of patients with idiopathic PAP [138], suggesting that GM-CSF is essential for maintaining pulmonary innate immunity.
It has been known for some time that GM-CSF can drive the differentiation of bone marrow progenitor cells into dendritic cells and macrophages [139,140]. Monocytes and DC precursors (pre-DCs) in the bone marrow are target progenitor cells of GM-CSF [141]. Recent studies have shown that GM-CSF-stimulated CCR2+ monocytes are critical for the pathogenesis of EAE [132,142]. Mice with Csf2rb conditionally deleted in Ly6Chi CCR2+ monocytes are resistant to the development of EAE, which is phenocopied in the complete Csf2rb knockout [132], suggesting that even though GM-CSF can act on many immune cell types, its effects on monocytes are the most critical for driving neuroinflammation. Furthermore, using a transgenic mouse line in which GM-CSF expression can be induced in peripheral CD4+ T cells, Spath et al. recently demonstrated that GM-CSF secretion promotes an antigen-independent invasion of inflammatory myeloid cell into the CNS, leading to tissue damage [143]. Myeloid cells invading into the CNS have a distinct genetic profile compared to those invading the lung, liver, and kidney [143]. The mechanism causing this difference remains to be determined.
Much effort has been made in the past few years to understand the mechanisms driving GM-CSF production in Th17 cells and its correlation with autoimmunity. Pertussis toxin-stimulated IL-1β production in myeloid cells promotes the expansion of GM-CSF-producing Th17 cells, which enhance their encephalitogenic potential [144]. IL-1β likely works upstream of IL-23 signaling, as the EAE pathogenicity of IL-1R1-deficient T cells is fully restored by the addition of exogenous IL-23 during in vitro polarization and expansion [144]. In addition, mice lacking protein kinase CK2 show reduced Th17-cell development and attenuated expression of IL-17, GM-CSF, and IL-23 receptor [145]. Pharmacological inhibition of CK2 activity ameliorates EAE severity and relapse incidence [145]. Of note, however, Th lineages other than Th17 also produce GM-CSF. Sheng et al. showed that GM-CSF expression can be induced by IL-7-mediated signaling in CD4+ T cells independent of IFN-γ and IL-17 [146]. GM-CSF can also be induced in conventional αβ and γδ T cells under IL-12- or IL-23-stimulated conditions [147]. Surprisingly, GM-CSF expression in human Th cells is induced by IL-12 but is suppressed by IL-23 [148], indicating that there are distinct GM-CSF regulatory pathways in humans and mice. The diverse sources of GM-CSF during inflammation may explain why injections of anti-IL-12p40 mAb (ustekinumab) failed to improve clinical outcomes in patients with RRMS [149]. This result, although disappointing, further supports the notion that GM-CSF, but not IL-17 nor other Th17 cell-producing cytokines, drives autoimmunity in the CNS.
Recently, the first drug for treating progressive MS, ocrelizumab, has been approved by the United States Food and Drug Administration. Ocrelizumab is a humanized anti-CD20 mAb which depletes non-antibody-secreting, CD20-expressing B cells [150]. This positive result suggests that B cells have regulatory role(s) that contributes to the pathogenesis of progressive MS, and that regulatory role may be involved in the production of GM-CSF [151]. Therefore, targeting GM-CSF or its downstream signaling may have potential in treating progressive MS.
8. Conclusions
Both clinical data and EAE model have provided strong evidence that targeting GM-CSF or GM-CSFR is a promising strategy for treating MS, and research in this area is underway. A recent study demonstrated that blocking GM-CSF signaling by anti-GM-CSFRα antibody results in amelioration of EAE progression [152]. In addition, a randomized phase 1b trial of anti-GMCSF mAb (MOR103) in patients with RRMS or SPMS has proven its tolerability [153] and therefore warrants further efficacy studies. However, although immunotherapy utilizing monoclonal antibodies has been proven successful in many disease settings, antibodies targeting GM-CSF or GM-CSFR have to be used with caution. As discussed above, GM-CSF is critical for the terminal differentiation of AMs via PU.1 [135], and GM-CSF autoantibodies are detected in patients with PAP [138]. Complete inhibition of GM-CSF signaling therefore may lead to lung pathology. Thus, mechanistic studies are needed to further distinguish signaling pathways responsible for driving immune cell development and functions under physiological versus pathological conditions, so that partial ablation of GM-CSF activity by targeting its downstream signaling molecules, such as the PU.1-independent pathway, may be feasible for achieving beneficial clinical outcomes.
Acknowledgments
This work was supported by NIH grant P20 GM109098 to Edwin C.K. Wan.
Author Contributions
Pushpalatha Palle and Edwin Wan prepared the Table; Kelly Monaghan and Edwin Wan prepared the Figure; Pushpalatha Palle, Kelly Monaghan, Sarah Milne and Edwin Wan wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goldenberg, M.M. Multiple sclerosis review. Pharm. Ther. 2012, 37, 175–184. [Google Scholar]
- Bove, R.; Chitnis, T. Sexual disparities in the incidence and course of MS. Clin. Immunol. 2013, 149, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Castrop, F.; Haslinger, B.; Hemmer, B.; Buck, D. Review of the pharmacoeconomics of early treatment of multiple sclerosis using interferon beta. Neuropsychiatr. Dis. Treat. 2013, 9, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Tullman, M.J. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am. J. Manag. Care 2013, 19, S15–S20. [Google Scholar] [PubMed]
- Naci, H.; Fleurence, R.; Birt, J.; Duhig, A. Economic burden of multiple sclerosis: A systematic review of the literature. Pharmacoeconomics 2010, 28, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Loma, I.; Heyman, R. Multiple sclerosis: Pathogenesis and treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Kister, I.; Bacon, T.E.; Chamot, E.; Salter, A.R.; Cutter, G.R.; Kalina, J.T.; Herbert, J. Natural history of multiple sclerosis symptoms. Int. J. MS Care 2013, 15, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintore, M.; Frederiksen, J.L.; et al. MRI criteria for the diagnosis of multiple sclerosis: Magnims consensus guidelines. Lancet Neurol. 2016, 15, 292–303. [Google Scholar] [CrossRef]
- Wattjes, M.P.; Steenwijk, M.D.; Stangel, M. MRI in the diagnosis and monitoring of multiple sclerosis: An update. Clin. Neuroradiol. 2015, 25, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the mcdonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Simons, M. Chronic progressive multiple sclerosis—Pathogenesis of neurodegeneration and therapeutic strategies. Curr. Neuropharmacol. 2010, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Applebee, A.; Panitch, H. Early stage and long term treatment of multiple sclerosis with interferon-beta. Biologics 2009, 3, 257–271. [Google Scholar] [PubMed]
- Andersson, P.B.; Waubant, E.; Gee, L.; Goodkin, D.E. Multiple sclerosis that is progressive from the time of onset: Clinical characteristics and progression of disability. Arch. Neurol. 1999, 56, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Wolinsky, J.S.; Amato, M.P.; Comi, G. Evolving expectations around early management of multiple sclerosis. Ther. Adv. Neurol. Disord. 2010, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Group, P.R.S. The diagnosis of primary progressive multiple sclerosis. J. Neurol. Sci. 2003, 206, 145–152. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Kremenchutzky, M.; Rice, G.P.; Hader, W.; Baskerville, J.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study. 6. Applications to planning and interpretation of clinical therapeutic trials in primary progressive multiple sclerosis. Brain 1999, 122, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Richard-Miceli, C.; Criswell, L.A. Emerging patterns of genetic overlap across autoimmune disorders. Genome Med. 2012, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Bashinskaya, V.V.; Kulakova, O.G.; Boyko, A.N.; Favorov, A.V.; Favorova, O.O. A review of genome-wide association studies for multiple sclerosis: Classical and hypothesis-driven approaches. Hum. Genet. 2015, 134, 1143–1162. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Oksenberg, J.R. Genetic determinants of risk and progression in multiple sclerosis. Clin. Chim. Acta 2015, 449, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Alvarez, E. The immunopathophysiology of multiple sclerosis. Neurol. Clin. 2011, 29, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra274. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L. Tolerogenic dendritic cells induced by vitamin d receptor ligands enhance regulatory T cells inhibiting autoimmune diabetes. Ann. N. Y. Acad. Sci. 2003, 987, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, J.; Zheng, C.; Wu, J.; Cheng, Y.; Zhu, S.; Lin, C.; Cao, Q.; Zhu, J.; Jin, T. 1,25-dihydroxyvitamin d3-induced dendritic cells suppress experimental autoimmune encephalomyelitis by increasing proportions of the regulatory lymphocytes and reducing T helper type 1 and type 17 cells. Immunology 2017, 152, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, D.S.; Hygino, J.; Ferreira, T.B.; Kasahara, T.M.; Barros, P.O.; Monteiro, C.; Oliveira, A.; Tavares, F.; Vasconcelos, C.C.; Alvarenga, R.; et al. Vitamin d modulates different IL-17-secreting T cell subsets in multiple sclerosis patients. J. Neuroimmunol. 2016, 299, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Morales, V.H.; Fierros, G.; Lopez, R.L.; Martinez-Nava, G.; Flores-Aldana, M.; Flores-Rivera, J.; Hernandez-Giron, C. Vitamin d receptor gene polymorphisms are associated with multiple sclerosis in mexican adults. J. Neuroimmunol. 2017, 306, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Cierny, D.; Michalik, J.; Skerenova, M.; Kantorova, E.; Sivak, S.; Javor, J.; Kurca, E.; Dobrota, D.; Lehotsky, J. Apai, Bsmi and Taqi VDR gene polymorphisms in association with multiple sclerosis in Slovaks. Neurol. Res. 2016, 38, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Hughes, A.M.; Lay, M.L.; Ponsonby, A.L.; Dwyer, D.E.; Taylor, B.V.; Pender, M.P. Epstein-barr virus and multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist 2011, 17, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Odumade, O.A.; Hogquist, K.A.; Balfour, H.H., Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin. Microbiol. Rev. 2011, 24, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, J.D.; Kamradt, T.; Martin, R.; Munz, C. Epstein-barr virus: Environmental trigger of multiple sclerosis? J. Virol. 2007, 81, 6777–6784. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple sclerosis-a quiet revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.P.; Hartung, H.P.; Calabresi, P.A. Anti-alpha4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 2005, 64, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (fty720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Martin, R.; Jacobson, S. Chemokines in chronic progressive neurological diseases: Htlv-1 associated myelopathy and multiple sclerosis. J. Neurovirol. 1999, 5, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Brandstadter, R.; Katz Sand, I. The use of natalizumab for multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Kuchroo, V.K. Using eae to better understand principles of immune function and autoimmune pathology. J. Autoimmun. 2013, 45, 31–39. [Google Scholar] [CrossRef] [PubMed]
- McRae, B.L.; Vanderlugt, C.L.; Dal Canto, M.C.; Miller, S.D. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J. Exp. Med. 1995, 182, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Whitham, R.H.; Bourdette, D.N.; Hashim, G.A.; Herndon, R.M.; Ilg, R.C.; Vandenbark, A.A.; Offner, H. Lymphocytes from SJL/J mice immunized with spinal cord respond selectively to a peptide of proteolipid protein and transfer relapsing demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 1991, 146, 101–107. [Google Scholar] [PubMed]
- Mendel, I.; Kerlero de Rosbo, N.; Ben-Nun, A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2B mice: Fine specificity and T cell receptor V beta expression of encephalitogenic t cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Kerfoot, S.M.; Long, E.M.; Hickey, M.J.; Andonegui, G.; Lapointe, B.M.; Zanardo, R.C.; Bonder, C.; James, W.G.; Robbins, S.M.; Kubes, P. Tlr4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 2004, 173, 7070–7077. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, F.; Basso, C.; Preite, S.; Reboldi, A.; Baumjohann, D.; Perlini, L.; Lanzavecchia, A.; Sallusto, F. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1beta production by myeloid cells. Nat. Commun. 2016, 7, 11541. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific t cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J.M. Passive induction of experimental allergic encephalomyelitis. Nat. Protoc. 2006, 1, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of cd4 t cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, D.; Meshorer, A.; Hirshfeld, T.; Arnon, R.; Sela, M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur. J. Immunol. 1971, 1, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, D.; Webb, C.; Meshorer, A.; Arnon, R.; Sela, M. Protection against experimental allergic encephalomyelitis. Nature 1972, 240, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Avolio, C.; Livrea, P.; Trojano, M. Glatiramer acetate in multiple sclerosis: A review. CNS Drug Rev. 2007, 13, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase iii multicenter, double-blind placebo-controlled trial. The copolymer 1 multiple sclerosis study group. Neurology 1995, 45, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B.; et al. Extended use of glatiramer acetate (copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 multiple sclerosis study group. Neurology 1998, 50, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Jee, Y.; Piao, W.H.; Liu, R.; Bai, X.F.; Rhodes, S.; Rodebaugh, R.; Campagnolo, D.I.; Shi, F.D.; Vollmer, T.L. Cd4(+)cd25(+) regulatory t cells contribute to the therapeutic effects of glatiramer acetate in experimental autoimmune encephalomyelitis. Clin. Immunol. 2007, 125, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Stock, A.; Vainshtein, A.; Shezen, E.; Gal, H.; Friedman, N.; Arnon, R. Glatiramer acetate reduces th-17 inflammation and induces regulatory T-cells in the cns of mice with relapsing-remitting or chronic eae. J. Neuroimmunol. 2010, 225, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Begum-Haque, S.; Sharma, A.; Kasper, I.R.; Foureau, D.M.; Mielcarz, D.W.; Haque, A.; Kasper, L.H. Downregulation of IL-17 and IL-6 in the central nervous system by glatiramer acetate in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2008, 204, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Meshorer, A.; Sela, M.; Arnon, R. Oral treatment of mice with copolymer 1 (glatiramer acetate) results in the accumulation of specific th2 cells in the central nervous system. J. Neuroimmunol. 2002, 126, 58–68. [Google Scholar] [CrossRef]
- Toker, A.; Slaney, C.Y.; Backstrom, B.T.; Harper, J.L. Glatiramer acetate treatment directly targets cd11b(+)ly6g(−) monocytes and enhances the suppression of autoreactive t cells in experimental autoimmune encephalomyelitis. Scand. J. Immunol. 2011, 74, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Herschkovitz, A.; Eilam, R.; Blumberg-Hazan, M.; Sela, M.; Bruck, W.; Arnon, R. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 11358–11363. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Domev, H.; Labunskay, G.; Sela, M.; Arnon, R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19045–19050. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A., Jr. Surface expression of alpha 4 integrin by cd4 t cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W.; et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Afzali, A.M.; Wiendl, H.; Meuth, S.G. Myelin oligodendrocyte glycoprotein (mog35–55) induced experimental autoimmune encephalomyelitis (EAE) in c57bl/6 mice. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Dornmair, K.; Meinl, E.; Wekerle, H. The search for the target antigens of multiple sclerosis, part 2: Cd8+ T cells, b cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016, 15, 317–331. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 353, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R. Natalizumab and progressive multifocal leucoencephalopathy. Ann. Rheum. Dis. 2006, 65, iii48–iii53. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, F.; Kajioka, J.; Miyamura, T. Prevalence rate and age of acquisition of antibodies against JC virus and BK virus in human sera. Microbiol. Immunol. 1982, 26, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Martin, R.; Sospedra, M. Long-term safety and efficacy of natalizumab in relapsing-remitting multiple sclerosis: Impact on quality of life. Patient Relat. Outcome Meas. 2014, 5, 25–33. [Google Scholar] [PubMed]
- Owens, G.P.; Gilden, D.; Burgoon, M.P.; Yu, X.; Bennett, J.L. Viruses and multiple sclerosis. Neuroscientist 2011, 17, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Calenoff, M.; Rozhon, E.; Lipton, H.L. Analysis of the complete nucleotide sequence of the picornavirus theiler’s murine encephalomyelitis virus indicates that it is closely related to cardioviruses. J. Virol. 1987, 61, 1507–1516. [Google Scholar] [PubMed]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Iwasaki, Y.; Terunuma, H.; Sako, K.; Ohara, Y. A comparative study of acute and chronic diseases induced by two subgroups of theiler’s murine encephalomyelitis virus. Acta Neuropathol. 1996, 91, 595–602. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and theiler’s virus-induced demyelinating disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar] [PubMed]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F.; Franklin, R.J. Remyelination in experimental models of toxin-induced demyelination. Adv. Mult. Scler. Expe. Demyelinating Dis. 2008, 318, 193–212. [Google Scholar]
- Torkildsen, O.; Brunborg, L.A.; Myhr, K.M.; Bo, L. The cuprizone model for demyelination. Acta Neurol. Scand. 2008, 188, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced DE- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Van Engelen, B.G.; Pavelko, K.D.; Rodriguez, M. Enhancement of central nervous system remyelination in immune and non-immune experimental models of demyelination. Mult. Scler. J. 1997, 3, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Maimone, D.; Gregory, S.; Arnason, B.G.; Reder, A.T. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J. Neuroimmunol. 1991, 32, 67–74. [Google Scholar] [CrossRef]
- Ruddle, N.H.; Bergman, C.M.; McGrath, K.M.; Lingenheld, E.G.; Grunnet, M.L.; Padula, S.J.; Clark, R.B. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 1990, 172, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.R.; Zaczynska, E.; Katsetos, C.D.; Platsoucas, C.D.; Oleszak, E.L. Differential expression of TGF-beta, IL-2, and other cytokines in the cns of theiler’s murine encephalomyelitis virus-infected susceptible and resistant strains of mice. Virology 2000, 278, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Koh, C.S.; Yahikozawa, H.; Yanagisawa, N.; Yagita, H.; Ishihara, Y.; Kim, B.S. The level of tumor necrosis factor-alpha producing cells in the spinal cord correlates with the degree of theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int. Immunol. 1996, 8, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The lenercept multiple sclerosis study group and the university of british columbia MS/MRI analysis group. Neurology 1999, 53, 457–465.
- Figiel, I. Pro-inflammatory cytokine TNF-alpha as a neuroprotective agent in the brain. Acta Neurobiol. Exp. (Wars) 2008, 68, 526–534. [Google Scholar] [PubMed]
- Jacobs, L.; O’Malley, J.; Freeman, A.; Ekes, R. Intrathecal interferon reduces exacerbations of multiple sclerosis. Science 1981, 214, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.; Salazar, A.M.; Herndon, R.; Reese, P.A.; Freeman, A.; Josefowicz, R.; Cuetter, A.; Husain, F.; Smith, W.A.; Ekes, R.; et al. Multicentre double-blind study of effect of intrathecally administered natural human fibroblast interferon on exacerbations of multiple sclerosis. Lancet 1986, 2, 1411–1413. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hirsch, R.L.; Haley, A.S.; Johnson, K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Wang, D.; Ghosh, D.; Islam, S.M.; Moorman, C.D.; Thomason, A.E.; Wilkinson, D.S.; Mannie, M.D. IFN-beta facilitates neuroantigen-dependent induction of cd25+ foxp3+ regulatory T cells that suppress experimental autoimmune encephalomyelitis. J. Immunol. 2016, 197, 2992–3007. [Google Scholar] [CrossRef] [PubMed]
- Ramgolam, V.S.; Sha, Y.; Jin, J.; Zhang, X.; Markovic-Plese, S. Ifn-beta inhibits human Th17 cell differentiation. J. Immunol. 2009, 183, 5418–5427. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.P.; Ma, D.H.; Wei, L.; van der Meide, P.H.; Mix, E.; Zhu, J. IFN-beta suppresses experimental autoimmune neuritis in lewis rats by inhibiting the migration of inflammatory cells into peripheral nervous tissue. J. Neurosci. Res. 1999, 56, 123–130. [Google Scholar] [CrossRef]
- Pennell, L.M.; Fish, E.N. Interferon-beta regulates dendritic cell activation and migration in experimental autoimmune encephalomyelitis. Immunology 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, S.; Cheng, G.; Guo, B. Type i ifn promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS ONE 2011, 6, e28432. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory b cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Streeter, H.B.; Rigden, R.; Martin, K.F.; Scolding, N.J.; Wraith, D.C. Preclinical development and first-in-human study of atx-ms-1467 for immunotherapy of ms. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e93. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q.; Wittmer, S.; Dalton, D.K. Failure to suppress the expansion of the activated cd4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted ifn-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 1996, 156, 5–7. [Google Scholar] [PubMed]
- Krakowski, M.; Owens, T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 1996, 26, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Pavelko, K.; Coffman, R.L. Gamma interferon is critical for resistance to theiler’s virus-induced demyelination. J. Virol. 1995, 69, 7286–7290. [Google Scholar] [PubMed]
- Hsieh, C.S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of th1 cd4+ T cells through IL-12 produced by listeria-induced macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Manetti, R.; Parronchi, P.; Giudizi, M.G.; Piccinni, M.P.; Maggi, E.; Trinchieri, G.; Romagnani, S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 1993, 177, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Durell, B.G.; Noelle, R.J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Investig. 2002, 110, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Gran, B.; Zhang, G.X.; Yu, S.; Li, J.; Chen, X.H.; Ventura, E.S.; Kamoun, M.; Rostami, A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: Evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J. Immunol. 2002, 169, 7104–7110. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic t cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing cd4+ effector t cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(h)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory t cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(h)17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic th17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(h)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine gm-csf. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. Rorgammat drives production of the cytokine gm-csf in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- McQualter, J.L.; Darwiche, R.; Ewing, C.; Onuki, M.; Kay, T.W.; Hamilton, J.A.; Reid, H.H.; Bernard, C.C. Granulocyte macrophage colony-stimulating factor: A new putative therapeutic target in multiple sclerosis. J. Exp. Med. 2001, 194, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, P.B.; Provitera, V.; De Rosa, T.; Tartaglia, G.; Gorga, F.; Perrella, O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: A correlation with clinical activity. Immunopharmacol. Immunotoxicol. 1998, 20, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.J.; Khademi, M.; Aram, J.; Ammann, S.; Kockum, I.; Constantinescu, C.; Gran, B.; Piehl, F.; Olsson, T.; Codarri, L.; et al. Multiple sclerosis-associated IL2ra polymorphism controls gm-csf production in human th cells. Nat. Commun. 2014, 5, 5056. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, J.; Ciric, B.; Imitola, J.; Gonnella, P.; Hwang, D.; Mahajan, K.; Mari, E.R.; Safavi, F.; Leist, T.P.; Zhang, G.X.; et al. Expression of gm-csf in t cells is increased in multiple sclerosis and suppressed by ifn-beta therapy. J. Immunol. 2015, 194, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, M.A.; Anderson, D.; Cerretti, D.P.; Price, V.; McKereghan, K.; Tushinski, R.J.; Mochizuki, D.Y.; Larsen, A.; Grabstein, K.; Gillis, S.; et al. Cloning, sequence, and expression of a human granulocyte/macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA 1985, 82, 6250–6254. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.M.; Gough, J.; Metcalf, D.; Kelso, A.; Grail, D.; Nicola, N.A.; Burgess, A.W.; Dunn, A.R. Molecular cloning of cdna encoding a murine haematopoietic growth regulator, granulocyte-macrophage colony stimulating factor. Nature 1984, 309, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.; et al. Granulocyte-macrophage colony-stimulating factor (gm-csf) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Sato, N.; Arai, K.; Miyajima, A. Expression cloning of the human IL-3 receptor cdna reveals a shared beta subunit for the human IL-3 and gm-csf receptors. Cell 1991, 66, 1165–1174. [Google Scholar] [CrossRef]
- Quelle, F.W.; Sato, N.; Witthuhn, B.A.; Inhorn, R.C.; Eder, M.; Miyajima, A.; Griffin, J.D.; Ihle, J.N. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol. Cell. Biol. 1994, 14, 4335–4341. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The cytokine gm-csf drives the inflammatory signature of ccr2+ monocytes and licenses autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by gm-csf: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.D.; Mueller, M.; Chou, T.H. Role of granulocyte/macrophage colony-stimulating factor in the regulation of murine alveolar macrophage proliferation and differentiation. J. Immunol. 1988, 141, 139–144. [Google Scholar] [PubMed]
- Shibata, Y.; Berclaz, P.Y.; Chroneos, Z.C.; Yoshida, M.; Whitsett, J.A.; Trapnell, B.C. Gm-csf regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001, 15, 557–567. [Google Scholar] [CrossRef]
- Suzuki, T.; Sakagami, T.; Rubin, B.K.; Nogee, L.M.; Wood, R.E.; Zimmerman, S.L.; Smolarek, T.; Dishop, M.K.; Wert, S.E.; Whitsett, J.A.; et al. Familial pulmonary alveolar proteinosis caused by mutations in csf2ra. J. Exp. Med. 2008, 205, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, U.; Nishinakamura, R.; Groneck, P.; Hattenhorst, U.; Nogee, L.; Murray, R.; Burdach, S. Human pulmonary alveolar proteinosis associated with a defect in gm-csf/IL-3/IL-5 receptor common beta chain expression. J. Clin. Investig. 1997, 100, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Tanaka, N.; Watanabe, J.; Uchida; Kanegasaki, S.; Yamada, Y.; Nakata, K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1999, 190, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Inaba, M.; Deguchi, M.; Hagi, K.; Yasumizu, R.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class ii-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. USA 1993, 90, 3038–3042. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Bottcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; Reis e Sousa, C. Gm-csf mouse bone marrow cultures comprise a heterogeneous population of cd11c(+)mhcii(+) macrophages and dendritic cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Brady, J.L.; Ryg-Cornejo, V.; Hansen, D.S.; Vremec, D.; Shortman, K.; Zhan, Y.; Lew, A.M. Gm-csf-responsive monocyte-derived dendritic cells are pivotal in th17 pathogenesis. J. Immunol. 2014, 192, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Spath, S.; Komuczki, J.; Hermann, M.; Pelczar, P.; Mair, F.; Schreiner, B.; Becher, B. Dysregulation of the cytokine gm-csf induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity 2017, 46, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Mufazalov, I.A.; Schelmbauer, C.; Regen, T.; Kuschmann, J.; Wanke, F.; Gabriel, L.A.; Hauptmann, J.; Muller, W.; Pinteaux, E.; Kurschus, F.C.; et al. IL-1 signaling is critical for expansion but not generation of autoreactive gm-csf+ th17 cells. EMBO J. 2017, 36, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Buhler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein kinase ck2 governs the molecular decision between encephalitogenic Th17 cell and treg cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.Y.; Kemeny, D.M.; Tan, P.; Moh, A.; Kaplan, M.H.; Zhang, Y.; et al. Stat5 programs a distinct subset of gm-csf-producing t helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Barr, M.J.; Chaplin, D.D.; Chi, H.; Kanneganti, T.D. Inflammasome-derived IL-1beta regulates the production of gm-csf by cd4(+) t cells and gammadelta T cells. J. Immunol. 2012, 188, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Noster, R.; Riedel, R.; Mashreghi, M.F.; Radbruch, H.; Harms, L.; Haftmann, C.; Chang, H.D.; Radbruch, A.; Zielinski, C.E. IL-17 and gm-csf expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 2014, 6, 241ra280. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H.; Ustekinumab, M.S.I. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008, 7, 796–804. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory gm-csf-producing b cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Davidson, T.S.; Kebir, H.; Xu, D.; Palacios-Macapagal, D.; Cann, J.; Rodgers, J.M.; Hunter, Z.N.; Pittet, C.L.; Beddow, S.; et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Asher, A.; Fryze, W.; Kozubski, W.; Wagner, F.; Aram, J.; Tanasescu, R.; Korolkiewicz, R.P.; Dirnberger-Hertweck, M.; Steidl, S.; et al. Randomized phase 1b trial of mor103, a human antibody to gm-csf, in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e117. [Google Scholar] [CrossRef] [PubMed]
- Teige, I.; Treschow, A.; Teige, A.; Mattsson, R.; Navikas, V.; Leanderson, T.; Holmdahl, R.; Issazadeh-Navikas, S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 2003, 170, 4776–4784. [Google Scholar] [CrossRef] [PubMed]
- Limmroth, V.; Putzki, N.; Kachuck, N.J. The interferon beta therapies for treatment of relapsing-remitting multiple sclerosis: Are they equally efficacious? A comparative review of open-label studies evaluating the efficacy, safety, or dosing of different interferon beta formulations alone or in combination. Ther. Adv. Neurol. Disord. 2011, 4, 281–296. [Google Scholar] [PubMed]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Prabhu Das, M.; Howard, E.D.; Weiner, H.L.; Sobel, R.A.; Kuchroo, V.K. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998, 161, 3299–3306. [Google Scholar] [PubMed]
- Havrdova, E.; Belova, A.; Goloborodko, A.; Tisserant, A.; Wright, A.; Wallstroem, E.; Garren, H.; Maguire, R.P.; Johns, D.R. Activity of secukinumab, an anti-IL-17a antibody, on brain lesions in rrms: Results from a randomized, proof-of-concept study. J. Neurol. 2016, 263, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.D.; Zehntner, S.P.; Kelly, L.M.; Bourbonniere, L.; Owens, T. Elevated interferon gamma expression in the central nervous system of tumour necrosis factor receptor 1-deficient mice with experimental autoimmune encephalomyelitis. Immunology 2006, 118, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Titelbaum, D.S.; Degenhardt, A.; Kinkel, R.P. Anti-tumor necrosis factor alpha-associated multiple sclerosis. Am. J. Neuroradiol. 2005, 26, 1548–1550. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).