In Vitro Efficient Expansion of Tumor Cells Deriving from Different Types of Human Tumor Samples


Abstract
:1. Introduction
2. Materials and Methods
2.1. Tumor Samples
2.2. Establishment of Primary Cell Cultures from Surgical Samples
2.3. Establishment of Primary Cell Cultures from Pleural or Peritoneal Effusions
2.4. Ascite-Derived Ovarian Cancer Cells
2.5. Phenotypical Analysis
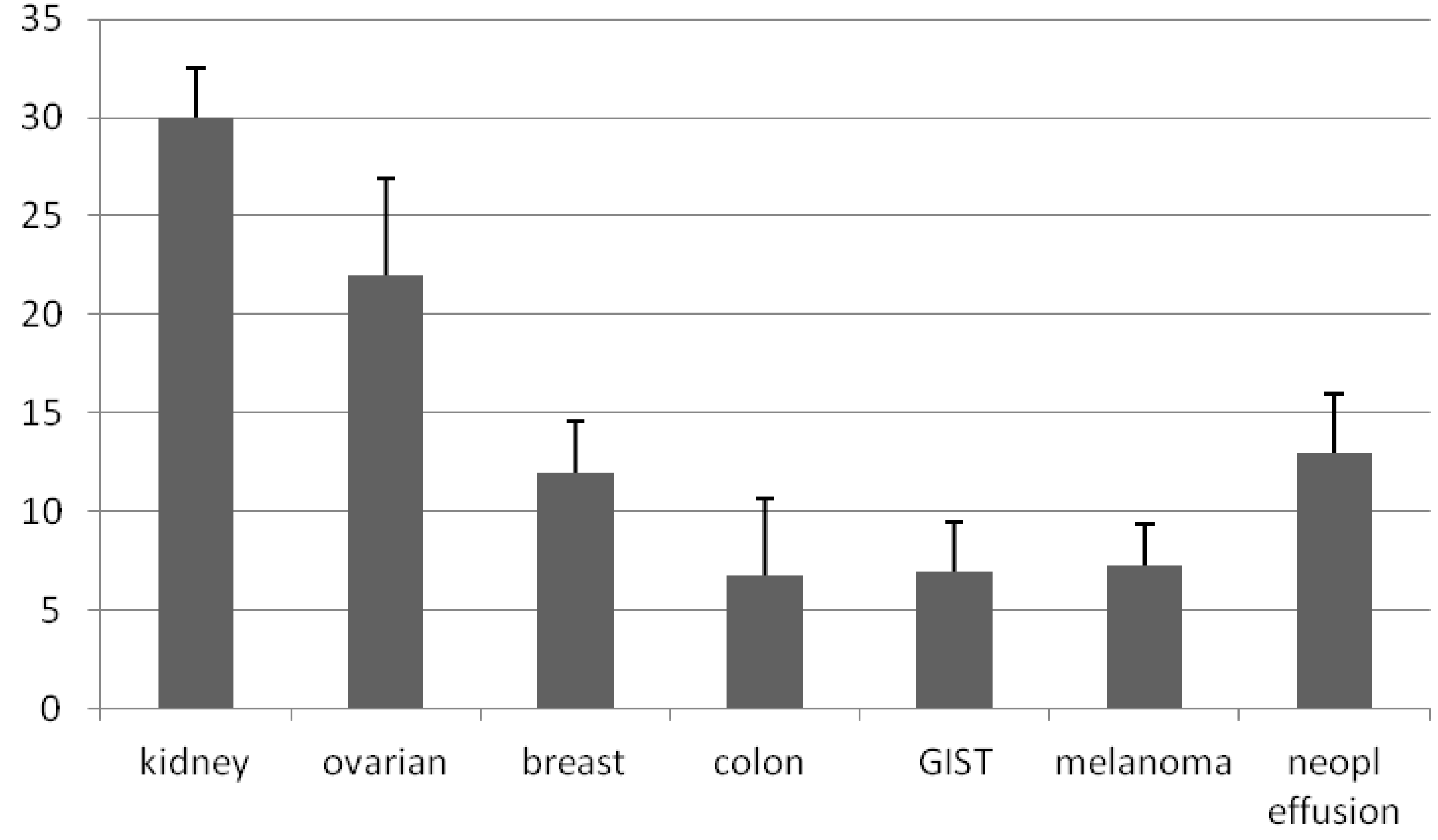
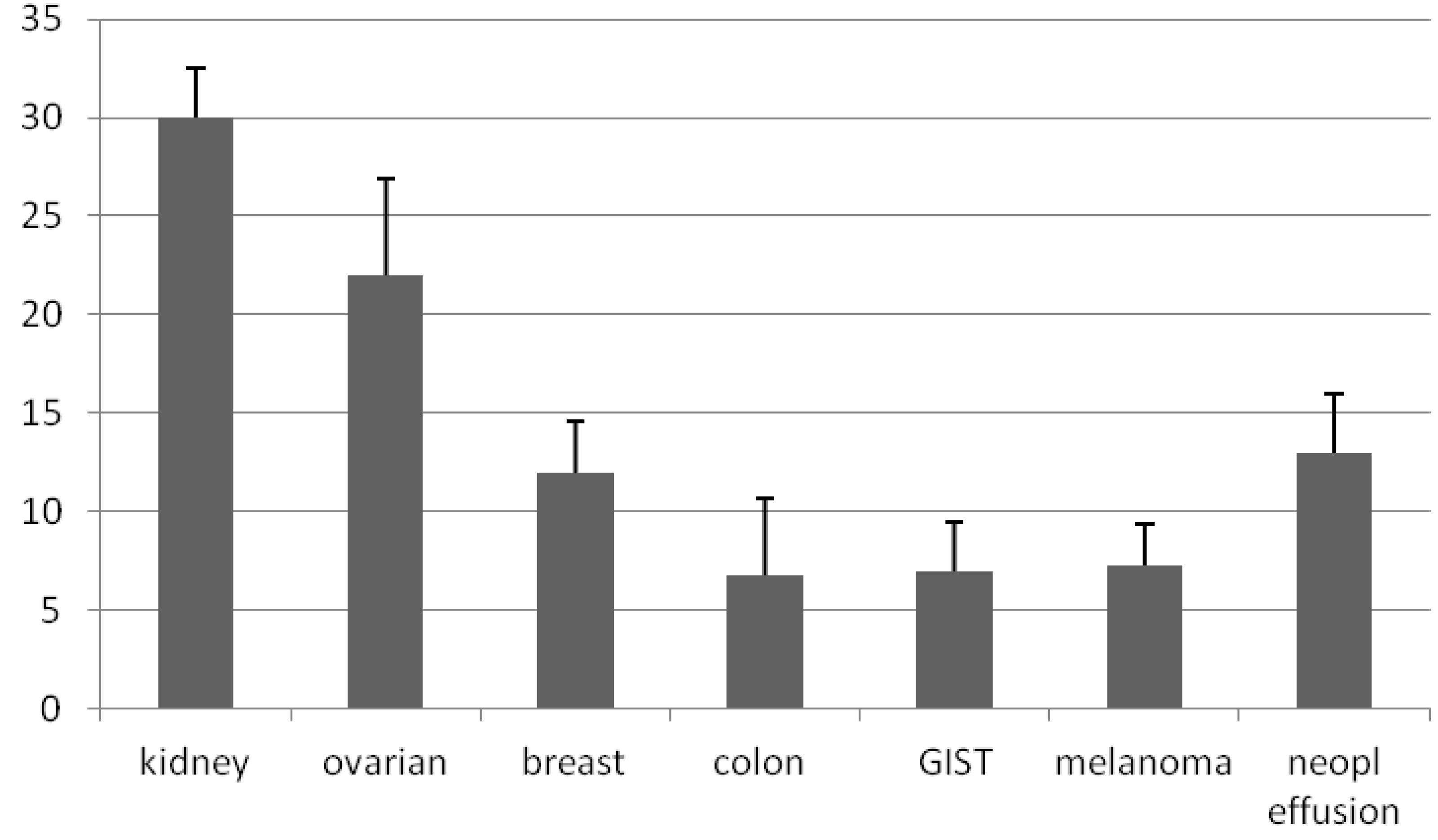
3. Results
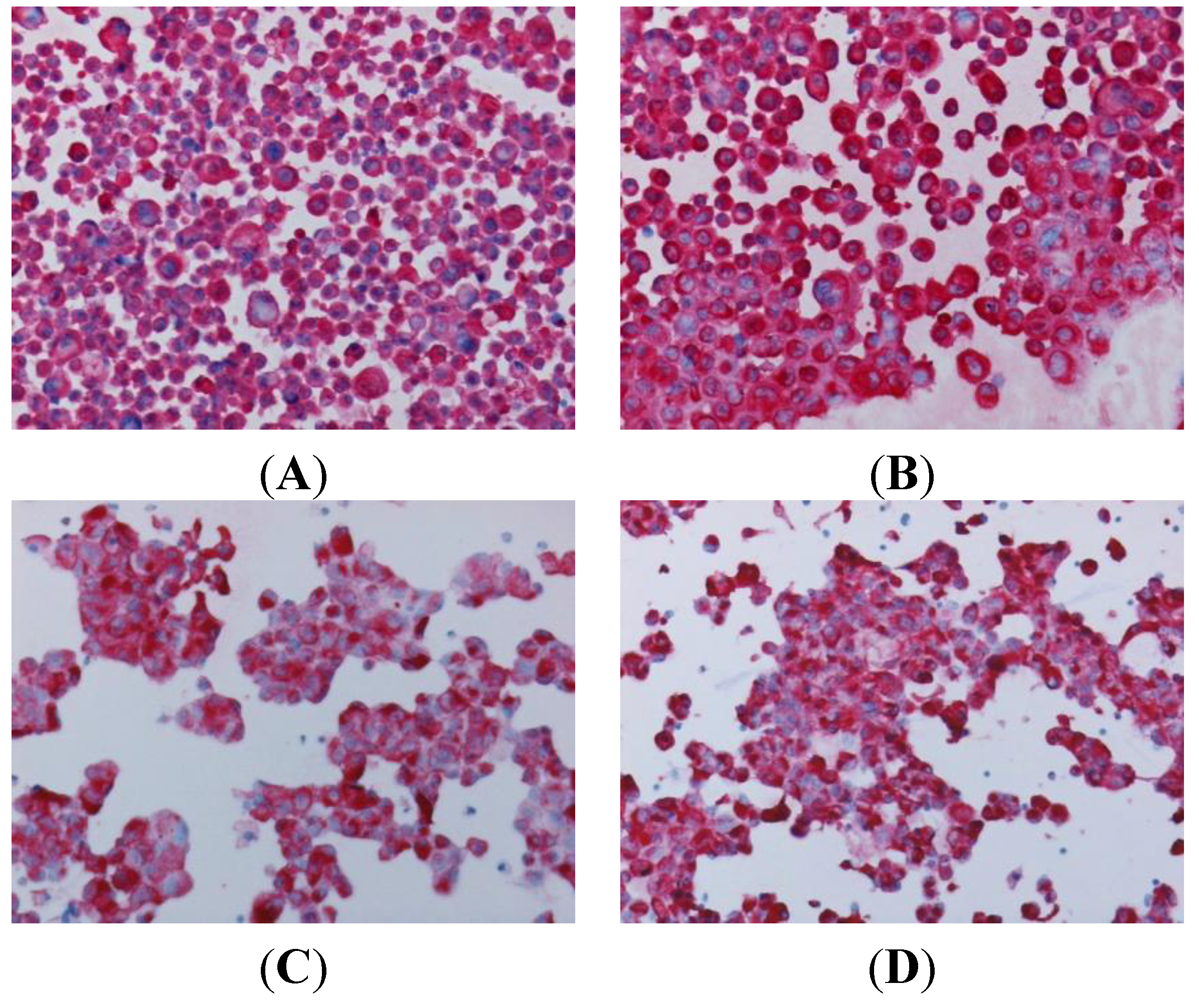
3.1. Generation of Primary Cultures

{kind=link}
{kind=link}
| Tumor type | Samples seeded | Tumor cell lines obtained | Success rate (%) |
|---|---|---|---|
| Kidney | 17 | 12 | 70.5 |
| Colon | 15 | 5 | 33.3 |
| Breast | 10 | 5 | 50 |
| Ovarian | 9 | 6 | 66.6 |
| Endometrial | 7 | 3 | 43 |
| Sarcoma | 6 | 4 | 66.6 |
| Melanoma | 6 | 2 | 33.3 |
| Gist * | 4 | 2 | 50 |
| Mesothelioma | 1 | 1 | 100 |
| Neoplastic effusion | 5 | 5 | 100 |
| Tumor type | Samples seeded | Tumor cell lines obtained | Success rate (%) |
|---|---|---|---|
| Kidney | 6 | 4 | 66 |
| Colon | 25 | 20 | 80 |
| Ovarian | 3 | 2 | 66.6 |
| Sarcoma | 6 | 5 | 83.3 |
3.2. Ascite-Derived Ovarian Cancer Cells
| Patients | Before purification | After purification | * Tumor cell lines | ¶ Tumor cells cryopreserved | ||
|---|---|---|---|---|---|---|
| Cell count | % CD326+ | Cell count | % CD326+ | |||
| AF001 | 30 × 106 | 3 | 5 × 104 | 70 | ND ‡ | ND |
| AF002 | 30 × 106 | 70 | 15 × 106 | 85 | ND | 15 × 106 |
| AF003 | 30 × 106 | 82 | 20 × 106 | 90 | ND | 20 × 106 |
| AF004 | 150 × 106 | 5 | 3 × 106 | 85 | ND | 3 × 106 |
| AF005 | 3 × 106 | 2 | 1 × 104 | 80 | yes | 2 × 106 |
| AF006 | 25 × 106 | 66 | 15 × 106 | 90 | ND | 15 × 106 |
| AF007 | 82 × 106 | 4 | 2 × 106 | 88 | ND | 2 × 106 |
| AF008 | 30 × 106 | 85 | 20 × 106 | 95 | ND | 20 × 106 |
| AF009 | 13 × 106 | 4 | 30 × 104 | 75 | yes | 4 × 106 |
| AF0010 | 10 × 106 | 50 | 12 × 106 | 95 | ND | 12 × 106 |
3.3. Characterization of Primary Cultures

4. Discussion
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Ochs, R.L.; Fensterer, J.; Ohori, N.P.; Wells, A.; Gabrin, M.; George, L.D.; Kornblith, P. Evidence for the isolation, growth and characterization of malignant cells in primary cultures of human tumors. In Vitro Cell. Dev. Biol. Anim. 2003, 39, 63–79. [Google Scholar]
- Bonner-Weir, S.; Taneja, M.; Weir, G.C.; Tatarkiewicz, K.; Song, K.H.; Sharma, A.; O’Neil, J.J. In vitro cultivation of human islets from expanded ductal tissue. Proc. Natl. Acad. Sci. USA 2000, 97, 7999–8004. [Google Scholar] [CrossRef]
- Dairkee, S.H.; Paulo, E.C.; Traquina, P.; Moore, D.H.; Ljung, B.M.; Smith, H.S. Partial enzymatic degradation of stroma allows enrichment and expansion of primary breast tumor cells. Cancer Res. 1997, 57, 1590–1596. [Google Scholar]
- Croce, M.V.; Colussi, A.G.; Segal-Eiras, A. Assessment of methods for primary culture of human breast epithelia. J. Exp. Clin. Cancer Res. 1998, 17, 19–26. [Google Scholar]
- Chen, L.L.; Mann, E.; Greenberg, B. Removal of fibroblast from primary cultures of squamous cell carcinoma of the head and neck. J. Tissue Cult. Methods 1993, 15, 1–10. [Google Scholar] [CrossRef]
- Tveit, K.M.; Pihl, A. Do cell lines in vitro reflect the properties of the tumours of origin? A study of lines derived from human melanoma xenografts. Br. J. Cancer 1981, 44, 775–786. [Google Scholar] [CrossRef]
- Schiavo, R.; Tullio, C.; la Grotteria, M.; Andreotti, I.C.; Scarpati, B.; Romiti, L.; Bozzi, F.; Pedrazzoli, P.; Siena, S. Establishment and characterization of a new Ewing’s sarcoma cell line from a malignant pleural effusion. Anticancer Res. 2007, 27, 3273–3278. [Google Scholar]
- Montagna, D.; Schiavo, R.; Gibelli, N.; Pedrazzoli, P.; Tonelli, R.; Pagani, S.; Assirelli, E.; Locatelli, F.; Pession, A.; Fregoni, V.; et al. Ex vivo generation and expansion of anti-tumor cytotoxic T-cell lines derived from patients or their HLA-identical sibling. Int. J. Cancer 2004, 110, 76–86. [Google Scholar] [CrossRef]
- Parmiani, G.; de Filippo, A.; Novellino, L.; Castelli, C. Unique human tumor antigens: Immunobiology and use in clinical trials. J. Immunol. 2007, 178, 1975–1979. [Google Scholar]
- Hayashi, K.; Yonamine, K.; Masuko-Hongo, K.; Iida, T.; Yamamoto, K.; Nishioka, K. Clonal expansion of T cells that are specific for autologous ovarian tumor among tumor-infiltrating T cells in humans. Gynecol. Oncol. 1999, 74, 86–92. [Google Scholar] [CrossRef]
- Moritake, H.; Sugimoto, T.; Kuroda, H.; Hidaka, F.; Takahashi, Y.; Tsuneyoshi, M.; Yoshida, M.A.; Cui, Q.; Akiyoshi, K.; Izumi, T.; et al. Newly established Askin tumor cell line and over expression of focal adhesion kinase in Ewing sarcoma family of tumors cell lines. Cancer Genet. Cytogenet. 2003, 146, 102–109. [Google Scholar] [CrossRef]
- Behren, A.; Anaka, M.; Lo, P.H.; Vella, L.J.; Davis, I.D.; Catimel, J.; Cardwell, T.; Gedye, C.; Hudson, C.; Stan, R.; et al. The Ludwig Institute for Cancer research Melbourne Cell line panel. Pigment Cell Melanoma Res. 2013, 26, 597–600. [Google Scholar] [CrossRef]
- Hass, R.; Bertram, C. Characterization of human breast cancer epithelial cells (HBCEC) derived from long term cultured biopsies. J. Exp. Clin. Cancer Res. 2009, 28, 127. [Google Scholar] [CrossRef]
- Went, P.T.; Lugli, A.; Meier, S.; Bundi, M.; Mirlacher, M.; Sauter, G.; Dirnhofer, S. Frequent EpCam protein expression in human carcinomas. Hum. Pathol. 2004, 35, 122–128. [Google Scholar] [CrossRef]
- Turin, I.; Pedrazzoli, P.; Tullio, C.; Montini, E.; la Grotteria, M.C.; Schiavo, R.; Perotti, C.; Locatelli, F.; Carretto, E.; Maccario, R.; et al. GMP production of anti-tumor cytotoxic T-cell lines for adoptive T-cell therapy in patients with solid neoplasia. Cytotherapy 2007, 9, 499–507. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Montagna, D.; Turin, I.; Schiavo, R.; Montini, E.; Zaffaroni, N.; Villa, R.; Secondino, S.; Schiavetto, I.; Caliogna, L.; Locatelli, F.; et al. Feasibility and safety of adoptive immunotherapy with ex-vivo generated autologous, cytotoxic T lymphocytes, in patients with solid tumors. Cytotherapy 2011, 14, 80–90. [Google Scholar]
- Secondino, S.; Turin, I.; Montini, E.; Porta, C.; Maccario, R.; Pedrazzoli, P.; Montagna, D. Case Report: Long lasting response in a patient with metastatic renal cell cancer receiving anti tumor cytotoxic T lymphocytes. Tumori 2013, 99, 282e–284e. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Turin, I.; Schiavo, R.; Maestri, M.; Luinetti, O.; Bello, B.D.; Paulli, M.; Dionigi, P.; Roccio, M.; Spinillo, A.; Ferulli, F.; et al. In Vitro Efficient Expansion of Tumor Cells Deriving from Different Types of Human Tumor Samples. Med. Sci. 2014, 2, 70-81. https://doi.org/10.3390/medsci2020070
Turin I, Schiavo R, Maestri M, Luinetti O, Bello BD, Paulli M, Dionigi P, Roccio M, Spinillo A, Ferulli F, et al. In Vitro Efficient Expansion of Tumor Cells Deriving from Different Types of Human Tumor Samples. Medical Sciences. 2014; 2(2):70-81. https://doi.org/10.3390/medsci2020070
Chicago/Turabian StyleTurin, Ilaria, Roberta Schiavo, Marcello Maestri, Ombretta Luinetti, Barbara Dal Bello, Marco Paulli, Paolo Dionigi, Marianna Roccio, Arsenio Spinillo, Federica Ferulli, and et al. 2014. "In Vitro Efficient Expansion of Tumor Cells Deriving from Different Types of Human Tumor Samples" Medical Sciences 2, no. 2: 70-81. https://doi.org/10.3390/medsci2020070
APA StyleTurin, I., Schiavo, R., Maestri, M., Luinetti, O., Bello, B. D., Paulli, M., Dionigi, P., Roccio, M., Spinillo, A., Ferulli, F., Tanzi, M., Maccario, R., Montagna, D., & Pedrazzoli, P. (2014). In Vitro Efficient Expansion of Tumor Cells Deriving from Different Types of Human Tumor Samples. Medical Sciences, 2(2), 70-81. https://doi.org/10.3390/medsci2020070